Где лечат мышечную дистрофию. Мышечная дистрофия лечение. Два важных вывода
Мышечная дистрофия входит в группу заболеваний, вызывающих дегенерацию скелетных мышц и суставов. Препаратов для его лечения пока не существует, хотя есть эффективные медицинские процедуры, способные замедлить развитие заболевания. Симптомы могут быть слабо выраженными, но в тяжелых случаях болезнь ухудшает рефлексы и лишает больного способности ходить.
Разновидности мышечной дистрофии
Выявлено более 30 видов мышечной дистрофии, из них наиболее распространены 9:
- миодистрофия Дюшенна – наиболее распространенная форма, развивается преимущественно у молодых мужчин;
- дистрофия Беккера – к сорокалетию больной теряет способность самостоятельно передвигаться;
- миотония – симптомы проявляются чаще у взрослых (либо у детей в период позднего детства);
- врожденная дистрофия – диагностируется сразу после рождения или в первые месяцы жизни;
- лимб-поясная – свойственна подросткам, молодым людям (20-25 лет), затрагивает мышцы бедер и плеч;
- плече-лопаточно-лицевая – вызывает ослабление мышц лица, плечевых мышц, теряется способность поднимать руки, возникают проблемы с речью, тахикардия;
- дистальная – для заболевания характерна слабость рук и нижних конечностей;
- дистрофия Эмери-Дрейфуса – для женщин опасна параличом сердца, у детей (преимущественно мальчиков), кроме сердца, поражаются икроножные мышцы и верхний плечевой пояс;
- болезнь Шарко (и прочие дегенеративные заболевания) также могут провоцировать схожие с мышечной дистрофией симптомы.
Из этих 9 форм мышечной дистрофии наиболее часто диагностируют следующие:
- Дистрофия Дюшенна. Симптомы проявляются в раннем детстве (до 5 лет). Болезнь диагностируют у одного из 3500 новорожденных мальчиков. Это наиболее распространенное смертельное генетическое расстройство. Женщины могут быть носителями гена, подвергшегося мутации, но сами этим расстройством не страдают. Дегенеративные изменения блокируют белок (дистрофин), поддерживающий структурные элементы мышечной ткани и клеточных мембран. Без дистрофина мышечная слабость прогрессирует, провоцируя обездвиженность и смерть.
- Дистрофия Беккера. Хотя производство дистрофина при этом заболевании сохраняется, уровень стержневидного белка существенно ниже нормы. Мышечная дистрофия развивается к 12-летнему возрасту. Болезнь развивается медленнее, но поражения значительны: искривление позвоночника, трудности с дыханием, постоянная усталость, слабость, болезни сердца, когнитивные проблемы.
Основные симптомы дистрофии
Хотя все расстройства связаны между собой, каждый тип дистрофии вызван уникальной мутацией генов, поэтому симптомы и время проявления заболевания разнятся:
- потеря мышечной массы (проблема усугубляется с возрастом);
- поражение нижних конечностей (далее слабость распространяется на мышцы шеи, плечи, спину, грудь);
- прогрессирующее истощение;
- увеличение икроножных и дельтовидных мышц;
- снижение гибкости, выносливости, усталость;
- нарушение координации, синхронизации движений;
- падения (из-за потери координации и мышечной слабости);
- скованность суставов, боль при движении;
- замена фиброзной ткани мышечной;
- трудно приседать, наклоняться, подниматься по лестнице;
- аномальное развитие скелета (кривизна позвоночника, нарушение осанки);
- уменьшается диапазон движения (до полного паралича);
- развивается пневмония, другие проблемы с работой дыхательной системы, нарушается сердечная деятельность, что может привести к смерти (при дистрофии Дюшенна).
Дистрофия передается по наследству. Однако в 35% случаев заболевание провоцирует спонтанная мутация генов из-за нарушения выработки белка. Мужчины подвержены большему риску из-за наличия одной Х-хромосомы. Y-хромосома (ещё одна половая хромосома, имеющаяся у мужчин) не имеет копии гена дистрофина и на развитие мышечной дистрофии влияния не оказывает.

У женщин риск дистрофии уменьшается благодаря наличию двух Х-хромосом. Даже если одна из хромосом аномальна, здоровый ген может производить достаточно дистрофина. Вероятность передачи по наследству дефектного гена составляет 50%.
Диагностика мышечной дистрофии
При диагностике дистрофии проводят:
- физический экзамен – проверяется гибкость, тестируется мышечная сила, диапазон движения и другие характеристики пациента;
- электромиографию – исследуется электрическая активность внутри мышц;
- электрокардиограмму;
- биопсию мышц – позволяет установить, какой тип дистрофии развивается;
- магнитно-резонансную томографию – показывает, какие органы и ткани поражены.
Женщины могут пройти тест при планировании беременности, чтобы знать о наличии или отсутствии дефектного гена. По анализу крови можно определить наличие ферментов, связанных с аномальным развитием мышц.
Лечение мышечной дистрофии
Лечение зависит от типа дистрофии. Дистрофия Беккера и Дюшенна неизлечимы, однако есть средства, позволяющие управлять симптомами болезни. Чем больше методик комбинируется (включая диету, эмоциональную и физиотерапию, лекарственную поддержку), тем лучше результат.
Терапия может включать:
- прием стероидов (для борьбы с мышечной слабостью и снятия болевых симптомов), альбутерола (при наличии астмы), лекарств, улучшающих работу сердца (ангиотензина, бета-блокаторов), ингибиторов протонной помпы, биодобавок, позволяющих поддерживать энергию и мышечную массу;
- использование ортопедических изделий (скобы, инвалидные кресла);
- лечение патологии речи (если затронуты лицо и язык).

Естественные методы управления мышечной дистрофией
Физическая активность
Важно пытаться сохранить гибкость и силу мышц. Пассивный режим, отсутствие элементарных нагрузок усугубляют симптомы болезни, способствуют развитию осложнений и эмоционального расстройства. Для поддержания гибкости и координации тела полезна физиотерапия. Мобильность можно повысить, используя ортопедические вкладки, корсет, трость, электроскутер, коляску, можно двигаться при поддержке ассистента.
От боли в суставах, для улучшения балансировки, уменьшения тревожности, сохранения диапазона движений полезно плавание, йога, упражнения на растяжку (для страдающих мышечной дистрофией разработаны специальные практики, найти их можно в интернете или у врача).
Психологическая поддержка
Здоровая диета
Некоторые пищевые химические вещества воздействуют на геном человека и меняют структуру генов, провоцируя возникновение или прогрессирование хронических заболеваний, развитие осложнений. Воспалительные процессы поддерживает обедненная полезными веществами (или перегруженная обработанными продуктами) диета, отсутствие физической активности, стресс. Эти факторы снижают способность организма защищать клетки от дегенерации, ускоряют старение, приводят к потере плотности костной ткани.
Противовоспалительная диета хоть и не сможет исцелить от мышечной дистрофии, но существенно замедлит прогрессирование болезни. Такой рацион уменьшает воспаления, подщелачивает организм, снижает уровень глюкозы, помогает вывести токсины, обеспечивает питательными веществами.
Принципы питания:
- замена «плохих» жиров «хорошими» – отказ от гидрогенизированного масла (соевого, рапсового), трансжиров, введение полезных насыщенных и ненасыщенных жиров (оливкового, подсолнечного, кокосового, льняного, кунжутного масла, авокадо, омега-3 жиров);
- выбор органических мясопродуктов (без антибиотиков, гормонов);
- исключение рафинированного сахара и глютена.
Лучшие противовоспалительные продукты — листовые овощи, китайская капуста, сельдерей, свекла, брокколи, черника, ананас, лосось (дикий), костный бульон, грецкие орехи, куркума, имбирь, семена чиа.

Кроме чистой воды, полезны травяные чаи, свежевыжатые соки, натуральный лимонад, морс, квас. Из молочных продуктов допустимо оставить в меню козье молоко (сыр, йогурт из него), овечий сыр.
Важно исключить вероятность попадания в организм токсинов не только из пищи, но и из бытовой химии, косметики, выбирая натуральные средства.
Добавки, поддерживающие мышечную ткань
По назначению врача, при мышечной дистрофии можно использовать следующие биодобавки:
- аминокислоты (карнитин, коэнзим Q10, креатинин) – способствуют выработке белка, необходимого для поддержки мышц;
- глюкозамин и хондроитин – борются с суставной болью;
- антиоксиданты (витамины С, Е, А) – полезны для сердца, суставов и мышц;
- зеленый, матча чай (экстракты) – поддерживают энергетический уровень;
- омега-3 кислоты, рыбий жир – противостоят воспалениям;
- пребиотики – улучшают пищеварение.
Эфирные масла
Уменьшить отек, снять боль и другие симптомы, вызванные дегенерацией мышц, тканей и суставов, помогает масло мяты, ладана, имбиря, куркумы, мирры. Эффективно при лечении депрессии, для снятия тревожности масло лаванды, ромашки, грейпфрута, сандалового дерева. Можно добавлять масло по капле в диффузор увлажнителя воздуха, принимать на его основе ванны и использовать для массажа.
При обнаружении любых тревожных признаков (онемении лица, нарушении речи, походки, внезапной слабости, снижении гибкости) необходимо пройти обследование. Раннее вмешательство позволит затормозить развитие болезни и уменьшить негативное влияние симптомов мышечной дистрофии на качество жизни.
Мышечная дистрофия, или, как ее еще называют медики, миопатия — это заболевание генетической природы. В редких случаях оно развивает по внешним причинам. Чаще всего это наследственное заболевание, для которого характерны мышечная слабость, дегенерация мышц, уменьшение диаметра скелетных мышечных волокон, а в особо тяжелых случаях — мышечных волокон внутренних органов.
Что такое мышечная дистрофия?
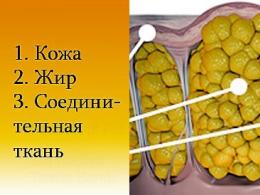
В процессе этого заболевания мышцы постепенно утрачивают способность к сокращению. Происходит постепенный распад. Мышечная ткань медленно, но неотвратимо заменяется жировой тканью и соединительными клетками.
Для прогрессирующей стадии характерны следующие симптомы мышечной дистрофии:
- сниженный болевой порог, а в некоторых случаях практические полная невосприимчивость к болевым ощущениям;
- мышечная ткань утратила свои способности к сокращению и росту;
- при некоторых разновидностях болезни — болевые ощущения в области мышц;
- атрофия скелетных мышц;
- неправильная походка вследствие недоразвития мышц ног, стоп вследствие неспособности выдерживать нагрузку при ходьбе;
- пациент часто желает присесть и полежать, так как у него попросту нет сил находиться на ногах — этот симптом характерен для больных женского пола;
- постоянная хроническая усталость;
- у детей — неспособность нормально учиться и усваивать новую информацию;
- изменение мышц в размерах — уменьшение в той или иной степени;
- постепенная утрата навыков у детей, дегенеративные процессы в психике у подростков.
Причины ее появления
Медицина до сих пор не может назвать все механизмы запуска мышечной дистрофии. Совершенно уверенно можно заявить одно: все причины лежат в изменении набора доминантных хромосом, которые отвечают в нашем организме за белковый и аминокислотный обмен. Без адекватного усвоения белка не будет нормального роста и функционирования мышц и костной ткани.

От вида хромосом, которые подверглись мутации, зависит течение болезни и ее форма:
- Мутация икс-хромосомы — распространенная причина мышечной дистрофии Дюшенна. Когда женщина-мать носит в себе такой поврежденный генный материал, можно говорить, что с вероятностью 70 % она передаст заболевание своим детям. При этом она зачастую патологиями мышечной и костной ткани не страдает.
- Миотоническая мышечная дистрофия проявляется по причине дефектного гена, принадлежащего девятнадцатой хромосоме.
- Половые хромосомы не влияют на локализацию мышечной недоразвитости: поясница-конечности, а также плечо-лопатка-лицо.
Диагностика заболевания
Диагностические мероприятия разнообразны. Существует множество недугов, напоминающих по тем или иным косвенными признакам проявления миопатии. Наследственность — самая частая причина заболевания «мышечная дистрофия». Лечение при этом возможно, но оно будет длительным и сложным. Обязательно нужно собрать сведения о режиме дня больного, об образе жизни. Как он питается, ест ли мясо и молочные продукты, употребляет ли алкогольные напитки или наркотики. Особенно важны эти сведения при диагностике мышечной дистрофии у подростков.

Подобные данные необходимы для составления плана проведения диагностических мер:
- электромиография;
- МРТ, компьютерная томография;
- биопсия мышечной ткани;
- дополнительная консультация ортопеда, хирурга, кардиолога;
- анализ крови (биохимия, общий) и мочи;
- соскоб мышечной ткани для анализа;
- генетическое тестирование для выявления наследственности пациента.
Разновидности заболевания
Исследуя на протяжении веков развитие прогрессирующей мышечной дистрофии, медики выявили следующие виды недуга:
- Дистрофия Беккера.
- Плече-лопаточно-лицевая мышечная дистрофия.
- Дистрофия Дюшенна.
- Врожденная мышечная дистрофия.
- Конечностно-поясная.
- Аутосомно-доминантная.
Это самые распространенные формы заболевания. Некоторые из них на сегодня успешно преодолимы благодаря развитию современной медицины. Некоторые имеют под собой наследственные причины, мутацию хромосом и терапии не поддаются.
Последствия недуга
Результат возникновения и прогресса миопатий различного генеза и этиологии — инвалидность. Серьезная деформация скелетных мышц и позвоночника приводит к частичной или полной утрате способности к движению.

Прогрессирующая мышечная дистрофия по мере своего развития часто приводит к развитию почечной, сердечной и дыхательной недостаточности. У детей — к умственному и физическому отставанию в развитии. У подростков — к нарушениям интеллектуальных и мыслительных способностей, остановке роста, карликовости, ухудшению памяти и потере способности к обучению.
Дистрофия Дюшенна
Это одна из самых тяжелых форм. Увы, современная медицина так и не смогла помочь больным прогрессирующей мышечной дистрофией Дюшенна адаптироваться к жизни. Большинство пациентов с этим диагнозом получают инвалидность с детства и не живут дольше тридцати лет.
Клинически проявляется уже в возрасте двух-трех лет. Дети не могут играть со сверстниками в подвижные игры, быстро утомляются. Часто наблюдается отставание в росте, в развитии речи и когнитивных функций. К пятилетнему возрасту мышечная слабость и недоразвитие скелета у ребенка становятся совершенно очевидными. Походка смотрится странно — слабые мышцы ног не позволяют больному идти ровно, не пошатываясь из стороны в сторону.
Родителям нужно начинать бить тревогу как можно раньше. Сделать как можно быстрее ряд генетических анализов, которые помогут с точностью установить диагноз. Современные методы лечения помогут больному вести приемлемый образ жизни, хотя и не восстановят полностью рост и функции мышечной ткани.
Дистрофия Беккера
Эта форма мышечной дистрофии была исследована Беккером и Кинером еще в 1955 году. В мире медицины ее именуют как мышечную дистрофию Беккера, или Беккера-Кинера.

Первичные симптомы такие же, как и у формы заболевания по Дюшенну. Причины развития так же и кроются в нарушении генного кода. Но в отличие от дистрофии Дюшенна, форма заболевания по Беккеру носит доброкачественный характер. Пациенты с этим типом болезни могут вести практически полноценную жизнедеятельность и дожить до преклонного возраста. Чем раньше диагностировать недуг и начать лечение, тем больше вероятность для пациента обычной человеческой жизни.
Не наблюдается и замедления развития умственных функций человека, свойственного злокачественной мышечной дистрофии по форме Дюшенна. При рассматриваемой болезни очень редко встречается кардиомиопатия и прочие отклонения в работе сердечно-сосудистой системы.
Плече-лопаточно-лицевая дистрофия
Эта форма заболевания достаточно медленно прогрессирует, имеет доброкачественный тип течения. Чаще всего первые проявления недуга заметны в возрасте шести-семи лет. Но иногда (примерно 15 % случаев) заболевание никак не проявляет себя до тридцати-сорока лет. В некоторых случаях (10 %) ген дистрофии не пробуждается вовсе в течение всей жизни пациента.
Как становится ясно из названия, поражаются мышцы лица, плечевого пояса и верхних конечностей. Отставание лопатки от спины и неровное положение уровня плеч, искривленная плечевая дуга — все это говорит о слабости или полной дисфункции передней зубчатой, трапециевидной и ромбовидной мышц. Со временем в процесс включаются двуглавые мышцы, задняя дельтовидная.

У опытного врача при взгляде на пациента может сложится обманчивое впечатление о наличии у него экзофтальма. Функция щитовидной железы при этом остается нормальной, обмен веществ чаще всего не затронут. Интеллектуальные возможности пациента также, как правило, сохранены. У больного есть все возможности вести полноценный, здоровый образ жизни. Визуально сгладить проявления плече-лопаточно-лицевой мышечной дистрофии помогут современные лекарственные препараты и физиотерапия.
Миотоническая дистрофия
Наследуется в 90 % случаев по аутосомно-доминантному типу. Поражает мышечную и костную ткань. Миотоническая дистрофия — очень редкое явление, частота его появления 1 к 10000, но эта статистика занижена, так как часто эта форма заболевания остается недиагностированной.
Дети, рожденные от матерей с миотонической дистрофией, часто страдают так называемой врожденной миотонической дистрофией. Она проявляется слабостью лицевых мышц. Параллельно часто наблюдается неонатальная дыхательная недостаточность, перебои в работе сердечно-сосудистой системы. Часто можно заметить отставание в умственном развитии, задержку психо-речевого развития у маленьких пациентов.
Врожденная мышечная дистрофия
В классических случаях гипотония заметна уже с младенческого возраста. Характерно уменьшение мышц и костной ткани в объеме наряду с контрактурами суставов рук и ног. В анализах активность сывороточного КК повышена. При биопсии пораженных мышц обнаруживается стандартная для мышечной дистрофии картина.

Эта форма носит не прогрессирующий характер, интеллект пациента почти всегда остается сохранным. Но, увы, многие больные врожденной формой мышечной дистрофии не могут передвигаться самостоятельно. Позднее может добавиться дыхательная недостаточность. Компьютерная томография иногда помогает выявить гипомиелинизацию слоев белого вещества мозга. Это не имеет известных клинических проявлений и чаще всего никак не сказывается на адекватности и психической состоятельности пациента.
Анорексия и психические расстройства как предвестники мышечного заболевания
Отказ многих подростков от приема пищи несет за собой необратимую дисфункцию мышечной ткани. Если в течение сорока дней в организм не попадают аминокислоты, не происходят процессы синтеза белковых соединений — мышечная ткань на 87 % отмирает. Поэтому родители должны следить за питанием детей, чтобы те не следовали новомодным анорексичным диетам. В рационе подростка должны ежедневно присутствовать мясо, молочные продукты и растительные источники белка.
В случаях запущенных расстройств пищевого поведения может наблюдаться полная атрофия некоторых мышечных участков, а в качестве осложнения часто появляется почечная недостаточность сначала в острой, а затем и в хронической форме.
Лечение и препараты
Дистрофия является серьезным хроническим заболеванием наследственного характера. Полностью ее вылечить невозможно, но современная медицина и фармакология позволяют откорректировать проявления недуга, чтобы сделать жизнь пациентов максимально комфортной.
Список препаратов, необходимых больным для лечения мышечной дистрофии:
- «Преднизон». Стероидное анаболическое средство, поддерживающее на высоком уровне синтез белка. При дистрофии позволяет сохранить и даже нарастить мышечный корсет. Является гормональным средством.
- «Дифенин» — также гормональный препарат со стероидным профилем. Имеет множество побочных эффектов и вызывает привыкание.
- «Оксандролон» — был разработан американскими фармацевтами специально для детей и женщин. Как и предшественники, является гормональным средством с анаболическим эффектом. Имеет минимум побочных эффектов, активно применяется для терапии в детском и юношеском возрасте.
- Гормон роста инъекционный — одно из новейших средств от мышечной атрофии и остановки роста. Очень эффективное средство, которое позволяет больным ничем не выделяться внешне. Для лучшего эффекта принимать его необходимо в детском возрасте.
- «Креатин» — натуральный и практически безопасный лекарственный препарат. Подходит детям и взрослым. Способствует росту мышц и предотвращает их атрофию, укрепляет костную ткань.
Мышечная дистрофия (МД) - это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются - теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Дистрофия или миопатия? Термин «миопатия» является общим названием для всех патологий М. Д. Мышечные дистрофии - это особые формы миопатий. Однако в повседневном языке термин миопатия часто используется для обозначения дегенерации мышц.
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:

Частота и тип заболеваний зависит от конкретной страны :

Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать - они теряют свой силовой потенциал и в результате атрофируются.
В ходе миопатии участвует несколько десятков разных генов. Чаще всего это гены, «производящие» белки, которые расположены в мембране мышечных клеток.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина - белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина - белка, составляющего мембрану клеток мышцы.
 Как и многие генетические заболевания, миопатия чаще всего передается родителями их ребенку. Реже эти заболевания могут «появляются» спонтанно, когда ген мутирует случайно. В этом случае больной ген отсутствует у родителей или других членов семьи.
Как и многие генетические заболевания, миопатия чаще всего передается родителями их ребенку. Реже эти заболевания могут «появляются» спонтанно, когда ген мутирует случайно. В этом случае больной ген отсутствует у родителей или других членов семьи.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
 Мышечная миопатия Дюшенна
. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Мышечная миопатия Дюшенна
. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера . Симптомы сравнимы с симптомами М. Д. Дюшенна, однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
 Миопатия Штейнтера
. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Миопатия Штейнтера
. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония - аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела . Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия . Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
 Врожденные МД
. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Врожденные МД
. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония . Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:

Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно - в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
 Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.

Болезнь мышечная дистрофия – целая группа похожих генетических заболеваний, которая характеризуется прогрессирующей атрофией (симметричной) скелетных мышц, которая в финальной стадии приводит к полной потере подвижности и смерти больного. Нервно-мышечная дистрофия особенно опасна для жизни, когда атрофия касается диафрагмы, а также межреберных мышц. Также нередки повреждения сердечной мышцы. При этом процесс может проходить неравномерно, захватывая только прилегающие к сухожилиям мышцы.
Такие болезни как прогрессирующая мышечная дистрофия протекают без потери чувствительности или болей в конечностях. Дистрофия мышечной ткани приводит к активному росту жировых клеток и соединительных тканей, создавая при этом ложное впечатление о нормальном состоянии мышц. Кроме того, может происходить частичный некроз мышечных волокон.
Врожденная мышечная дистрофия начинает проявляться уже в первые годы жизни ребенка – он отстает от сверстников в развитии моторики, поздно начинает садиться, вставать или ходить. Позже проявляются различные костно-суставные нарушения и деформация позвоночного столба и грудины.
Причины мышечной дистрофии
Мышечная дистрофия, причины которой кроются в генетическом дефекте, чаще проявляется у представителей мужского пола. У женщин поврежденный рецессивный ген компенсируется здоровым геном в Х-хромосоме.
Врожденная мышечная дистрофия характеризуется почти полным отсутствием необходимого для поддержания мышечной структуры белка дистрофина. При некоторых формах мышечной дистрофии белок вырабатывается, однако функционирует неправильно.
Симптомы мышечной дистрофии
Мышечная дистрофия, симптомы которой проявляются в равной степени у взрослых и детей, характеризуется заметным снижением мышечного тонуса, визуальным нарушением походки, что связано с атрофией скелетных мышц. У больных отсутствуют мышечные боли, но чувствительность при этом не нарушается. Мышечная дистрофия у детей часто приводит к утрате наработанных физических навыков, приобретенных до появления заболевания – ребенок не может ходить, не держит голову, перестает сидеть и т.д.
Прогрессирующая мышечная дистрофия приводит к увеличению мышц в объеме – места отмерших мышечных волокон занимает соединительная ткань. Больной часто падает и жалуется на постоянную усталость, полное отсутствие физических сил.
У детей мышечная дистрофия, причины которой кроются в генетических дефектах, может вызвать различные нейроповеденческие расстройства (гиперактивность, синдром дефицита внимания, аутизм в легкой форме).
Классификация мышечной дистрофии
- Болезнь мышечная дистрофия имеет несколько распространенных форм, среди которых:
- Мышечная дистрофия Дюшена (или псевдогипертрофическая) проявляется преимущественно у мальчиков в детском возрасте (первые симптомы появляются уже в возрасте 2-5 лет). Дистрофия мышечной ткани начинается с нижних конечностей и таза, затем поражает верхнюю половину тела вкупе с остальными группами мышц. Дегенерация приводит к заметному увеличению объема икроножных мышц. Растет количество жировой и соединительной ткани. Также увеличивается и ослабевает сердечная мышца. Мышечная дистрофия Дюшена, к сожалению, прогрессирует довольно быстро – к 12 ребенок теряет способность к движению, а к 20 – большинство пациентов умирает.
- Мышечная дистрофия Беккера развивается значительно медленнее, нежели предыдущая форма. Наиболее часто встречается у низкорослых людей. Мышечная дистрофия Беккера протекает таким образом, что пациенты довольно долго остаются в удовлетворительном состоянии. К инвалидности приводят только сопутствующие заболевания или травмы.
- Болезнь Штейнерта (миотоническая мышечная дистрофия) прогрессирует медленнее. Чаще встречается у взрослых пациентов 20-40 лет. Заболевание характеризуется миотонией (замедленное расслабление мышц), заметной слабостью мышц лица. Также возможно поражение и других групп мышечной ткани, к примеру, конечностей. Помимо скелетных, заболевание может поражать сердечную мышцу или мышцы внутренних органов.
- Юношеская форма или мышечная дистрофия Эрба начинается в 10-20 лет с атрофии мышечной ткани рук, плеч. Затем заболевание перекидывается на таз и ноги. Мышечная дистрофия Эрба протекает медленно. Характеризуется своеобразным "переваливанием" при ходьбе, пациенты ходят, выпячивая живот и отодвигая назад грудную клетку.
- Мышечная дистрофия Ландузи-Дежерина (эта форма заболевания носит название плече-лопаточно-лицевой) характеризуется поражением мышц лица и постепенной атрофией плечевого пояса, мышц туловища и конечностей. На ранних стадиях дегенерации мышечной ткани недостаточно смыкаются веки и губы, что в свою очередь приводит к нарушению дикции. Проявляется такая мышечная дистрофия у взрослых до 52 лет. Протекает очень медленно, что позволяет больному сохранять трудоспособность. Атрофия мышц таза начинается только через 15-25 лет, затрудняя движения.
Наиболее распространенным видом болезни мышечная дистрофия является прогрессирующая мышечная дистрофия.
Диагностика заболевания
Лечение мышечной дистрофии назначается исключительно после комплексной диагностики. Прогрессирующая мышечная дистрофия диагностируется врачами при проявлении у новорожденного нехарактерной мышечной слабости. В крови наблюдается повышенное содержание выделяемого из мышечных клеток фермента креатинкиназа.
Для уточнения диагноза производится биопсия мышечной ткани с последующими микроскопическими исследованиями, а также электромиография и измерение скорости нервных импульсов.
В случае предрасположенности обоих родителей к заболеванию проводится специальная перинатальная диагностика заболевания, позволяющая определить наличие дефектов гена у плода.
Как лечить мышечную дистрофию
Как лечить мышечную дистрофию, врачи ответить не могут. По сути, лечение мышечной дистрофии безуспешно. Предотвратить или даже замедлить темпы атрофии мышечной ткани на данном этапе развития медицины практически невозможно.
Прогрессирующая мышечная дистрофия, лечение которой направлено скорее на борьбу с осложнениями, чем на само заболевание, приводит к деформации позвоночника, частые пневмонии и проблемы с сердцем. Терапия позволяет заметно улучшить качество жизни пациентов.
В таких случаях лечение мышечной дистрофии включает в себя сразу несколько пунктов:
- пациент употребляет кортикостероиды, позволяющие облегчить некоторые симптомы болезни и добавляющие энергии;
- больным рекомендуются умеренные физические нагрузки, плаванье (полное бездействие приводит к ускорению дистрофии мышечной ткани);
- проводится специальная физиотерапия, направленная на поддержание мышечного тонуса и улучшение функциональности суставов, а также специальные дыхательные гимнастики;
- используются различные ортопедические приспособления (специальные стяжки для фиксации голени, инвалидные коляски и прочее).
Если прогрессирующая мышечная дистрофия (лечение и терапия могут быть крайне не эффективны) вызывает нарушения в работе диафрагмы и легочного аппарата, для поддержания нормального состояния пациента используются респираторные механизмы.
Мышечная дистрофия у детей
Такое генетическое заболевание как мышечная дистрофия у детей выражается неполнотой движений и недостаточной физической активностью. Это вызвано патологией скелетных мышц. Наиболее часто у детей проявляется прогрессирующая форма мышечной дистрофии, полученная ребенком в результате наследования пораженных генов.
Дистрофия мышечной ткани не видна сразу после рождения младенца. Она проявляется несколько позже, когда ребенок учится держать голову или делает первые самостоятельные движения.
Начинаясь с нарушений работы опорно-двигательного аппарата, заболевание приводит к инвалидности. А в финальной стадии – к летальному исходу.
Помочь детям с заболеванием мышечная дистрофия
На данный момент на попечении нашего фонда нет детей с данным диагнозом. Однако вы можете помочь больным детям с другими диагнозами!
СТАТЬИ ЭТОЙ ТЕМАТИКИ ДОЛЖНЫ ЧИТАТЬСЯ НЕ ТОЛЬКО БОЛЬНЫМИ МИОПАТИЕЙ, НО И ИХ РОДСТВЕННИКАМИ, БЛИЗКИМИ, ДРУЗЬЯМИ, ОДНОКЛАСНИКАМИ (ДАЖЕ В ПЕРВУЮ ОЧЕРЕДЬ). ЧТОБЫ ПОНИМАТЬ, ЧТО РУКА ПОМАГАЮЩАЯ БОЛЬНОМУ ДОЛЖНА БЫТЬ НЕ ТОЛЬКО СИЛЬНОЙ, НО И НЕЖНОЙ. НО И БРАТИКИ-МИОПАТИКИ ДОЛЖНЫ ВЕСТИ СЕБЯ ДОСТОЙНО (НУ ХОТЯ БЫ ПО ВОЗМОЖНОСТИ)!!!
Хайме Хиль, Антонио Альбарран
Мышечная дистрофия
Всех нас, и самих инвалидов, и их близких и друзей, беспокоят, как нынешние проблемы, так и проблемы, связанные с будущим. Особенно это относится к тем, кто страдает миопатией, иначе мышечной дистрофией, болезнью, которая является неизлечимой и прогрессирующей.
Усилить контроль за состоянием таких больных, разработать методы задержания хода болезни и добиться пусть частичного улучшения их здоровья — это, несомненно, благородная задача, ждущая своего решения.
Для начала полезно сделать обзор истории открытия мышечной дистрофии. Таким образом, узнав о том, что уже известно, мы будем иметь представление о современных исследованиях в области этой болезни и на что можем надеяться в будущем.
Глава 1. НЕМНОГО ИСТОРИИ
1. Причина в нервах
В течение долгих лет считалось, что разрушение мышц или прогрессирующая мышечная слабость происходит из-за недостаточного функционирования нервной системы. Согласно распространенным тогда теориям, пораженные нервы не способны активизировать мышечную ткань. До конца Х1Х века медики не допускали и мысли, что могут возникать болезни, которые нарушают нормальную деятельность мышц, не затрагивая нервы.
В то время были выявлены различные группы нервномышечных заболеваний. Многие из них, к сожалению, часто имели тяжелые последствия. Другие относительно менее тяжелые заболевания почти не прогрессировали. Некоторые поддавались лечению при помощи соответствующих медикаментозных средств.
В 1830 году шведский хирург Чарльз Белл дал первое описание болезни (клиническое), известной сейчас как мышечная дистрофия (МД), подтвердив, что причина ее в недостаточности неврогенетических функций (то есть нервного происхождения).
2. нервная или мышечная?
Позднее, в 1850 году, французский врач Франсуа Амилькар Аран привлек внимание медиков к целому ряду факторов, характеризирующих слабость и прогрессирующую атрофию мышц.
Два года спустя другой француз, Эдуар Мерьен, провел обследование четырех братьев, которые страдали ярко выраженным разрушением мышечной ткани. При обследовании он отметил, что мышцы были дегенерированы и полностью замещены скоплениями каких-то крупинок и жира, тогда как нервы и спинной мозг оставались без патологических изменений. Однако он, по непонятной причине, отказался от своего открытия и назвал болезнь н е р в н о — м ы ш е ч н о й а т р о ф и е й. Несмотря на явное противоречие такого диагноза и его несоответствие с данными исследований самого ученого, сделанное им заключение было признано другими исследователями как неоспоримое.
Почему же медики того времени продолжали утверждать, что эта болезнь происходит от нервов? Вероятно, единственная причина состояла в тогдашнем превосходстве школы французской неврологии, под началом Жана Мартэна Шарко. Мировая слава этого знаменитого клинициста сделала Париж «медицинской Меккой».
Несмотря на те скудные познания, которые имелись в области нарушений моторики (ведь еще не существовало понятия о рефлексах, не велись проводимости спинного мозга), в середине XIX века Шарко и его последователями были выделены в отдельную основную категорию все болезни, симптомами которых являлись разрушение или слабость мышц. И этой группе заболеваний они дали название «прогрессивный паралич».
3. Гениальный Дюшенн
Даже первооткрывателю в неврологии Г.Б. Аман Дюшенну не удалось избавиться от непреодолимого влияния идей Шарко.
Дюшенн описал в 1849 году одну из форм прогрессирующей нервно-мышечной атрофии, возникшую от недостаточности функций периферических нервов, — указывая, что болезнь началась с поражения рук и плечевого пояса, а затем постепенно распространялась на туловище и нижние конечности. В дальнейшей стадии больной мог умереть от какого-нибудь легочного заболевания в результате ослабления мышц дыхательной системы. Поскольку это сообщение Дюшенна было впоследствии опубликовано Араном, данная форма нервно-мышечной атрофии стала известна как Аран-Дюшенновская.
Дюшенн сделал большее. Он стал первым исследователем, открывшим метод электростимуляции мышц и других органов, исключающий прокалывание кожи электродами. Таким образом, одновременно стало возможным изучение сокращений мышц и различных других мышечных функций. Дюшенн также изобрел замечательный инструмент для взятия образцов мышечной оболочки.
4. Заболевание мышц
Итак, мы приближаемся к более точным сведениям об этой болезни.
В 1890 году немецкий врач Вильгельм Эрб опубликовал несколько монографий, где описывались сходные точки зрения на различные формы болезни, названной им «прогрессирующая мышечная дистрофия». Он указал на на четыре ее разновидности (три из которых появляются в детстве и подростковом периоде и одна — в зрелом возрасте) и снабдил текст рисунками деформаций, вызываемых каждой из них.
Эти четыре разновидности отличались друг от друга по таким признакам: по возрасту, в каком началась болезнь, по распространенности мышечного поражения и по тому, как быстро болезнь прогрессирует. Указанные разновидности обьединял один и тот же патологический процесс, то есть клиническая картина: начальный симптом ослабления мышц в области таза или плечевого пояса, характерная походка, наличие особых деформаций и прогрессирующее развитие болезни.
Эрб был убежден в том, что все это являлось следствием какого-то нарушения мышечного покрова.
В 1909 году Ф.А.Льевени и Л.Крайстлер, два североамериканских врача из Монтефьорской больницы Нью-Йорка открыли впервые биохимическое отклонение при мышечной дистрофии, а именно присутствие к р е а т и н а (производного аминокислот, которое накапливается в мышцах) в моче зачительно увеличивается после введения в желудок больного веществ, называемых протеинами. Исследования метаболизма креатина у больных мышечной дистрофией в дальнейшем проводились с тестом на усвояемость креатина. Этот тест применяется и теперь для определения болезней, связанных с поражением мышц. О нем мы поговорим позже.
5. Болезнь, передающаяся по наследству
Наследственный характер этого заболевания становится все более очевиден. Но в 1950 году доктор Ф.Г.Тейлер из Университета в Медицинском центре штата Юта точно установил наследственные законы при некоторых тяжелых случаях мышечной дистрофии. В этих немногих случаях, когда в истории семьи больного никто не страдал мышечной дистрофией, речь шла о появлении в ходе спонтанной мутации у самого пациента болезнетворных генов. Они могут обнаружиться у него при интенсивном облучении Х-лучами, при ядерном облучении, а также под воздействием других еще неизученных факторов.
6. На что можно надеяться?
Значение приведенных выше диагнозов в решение проблемы мышечной дистрофии в настоящее время нельзя недооценивать. Для человека с мышечной дистрофией, знающего, что его болезнь стабилизирована или развитие ее удается сдерживать, правильный крайне необходим. Многие больные очень часто сетуют на скудость информации об этом заболевании. Верный открыл бы такому человеку глаза на самого себя, дал бы ему надежду на лучшую жизнь. Даже если впереди трагическая развязка, замедленное течение болезни позволит человеку приспособиться к прогрессирующей инвалидности и развить множество возможностей, которые иначе могли бы остаться втуне.
За последние десятилетия исследования в области заболеваний мышц значительно расширились. Опыты на животных, достижения биохимии и генетики постоянно дают больше и больше сведений, помогающих лучше узнавать причины прогессирующей мышечной дистрофии. Некоторые думают, что мы находимся на пороге важных открытий. Нам не хотелось бы безответственно внушать кому-то надежду на «чудодейственное» лекарство, по которому вздыхает сейчас столько больных. Тем не менее будет справедливым надеяться, что лучшее знание причин болезни позволит найти средство для ее контроля и поддержания на определенной стадии, чтобы она не могла развиваться дальше, — если нельзя пока говорить о полном излечении.
7. Два важных вывода
Из краткого исторического обзора, который мы с вами только что совершили, можно сделать два вывода.
а) дистрофия имеет мышечное происхождение, то есть не является следствием недостаточности функции нервов, как считалось в течение многих лет, а происходит из-за повреждения мышечной оболочки.
b) прогрессирующая мышечная дистрофия передается в основном по наследству (только форма болезни под названием мышечная дистрофия позднего выявления, похоже, не связана с наследственностью). Об этой стороне проблемы поговорим в следующей главе.
Глава 2. МЫШЕЧНАЯ ДИСТРОФИЯ ЯВЛЯЕТСЯ НАСЛЕДСТВЕННОЙ
К такому заключению мы подошли в предыдущей главе после небольшого экскурса в историю. Оно, разумеется, не радует и воспринимается с трудом. Во-первых, потому что никому из нас не доставляет удовольствия признаваться в том, что в нашей семье есть те или другие проблемы. Во- вторых, потому что больные испытывают справедливую потребность продолжиться в детях, которые называли бы их папой и мамой, — но угроза произвести на свет новых дистрофиков ставит перед ними неразрешимую преграду.
В любом случае нам представляется возможным предложить некоторую информацию на эту тему, чтобы не прятать голову в песок (так делают страусы, когда хотят уйти от пугающей их действительности) и не давать больным повода к чрезмерному беспокойству.
Мы должны пояснить, что существуют два основных типа наследственности:
а) А в т о с о м н а я н а с л е д с т в е н н о с т ь, то есть, когда наследственные признаки передаются не половыми, а другими хромосомами. Так бывает, если являются носителями болезни, но сами при этом здоровы (в основном, они имеют здоровых детей, однако и в этих семьях бывают отдельные случаи появления заболевания). С помощью статистики можно определить, что каждый четвертый ребенок, независимо от пола, будет страдать этой болезнью. Такая передача очень обычна при большинстве форм дистрофии, кроме той, которая называется Дюшенновской. О ней будет сказано ниже.
b) П о л о в а я н а с л е д с т в е н н о с т ь. Речь идет о такой наследственности, при которой мать, оставаясь здоровой, является носительницей болезни. В соответствии со статистикой можно утверждать, что ее сыновья будут страдать дистрофией, а дочери также станут здоровыми носительницами болезни. Это обычная форма наследования детского паралича Дюшенна, или Дюшенновской болезни. Только в этом случае будет правильным часто повторяемое высказывание по поводу прогрессирующей мышечной дистрофии о том, что «механизм ее наследственности связан с полом».
1. Краткое пояснение
Наши читатели простят нам, что мы в своих объяснениях пользуемся довольно сложным языком. Нам хотелось бы давать их другими, более простыми словами, но это не представляется возможным. Поэтому тем, кому предназначена эта книга, потребуется небольшое усилие. Итак, не пора ли нам углубиться в суть темы?
Постараемся понять, как происходит процесс размножения.
В каждой клетке существуют две серии хромосом, которые образуют гомологические пары. Эти две — а они могут быть одинаковыми по форме и размерам — названы половыми, тогда как остальные пары гомологических хромосом, которые идентичны и у самки и у самца, получили название автосом. Так например, в клетках человека содержится двадцать две пары автосом и лишь одна пара половых хромосом.
У самок млекопитающих (человек, как известно, тоже принадлежит к этому классу) половые хромосомы идентичны и называются хромосомами Икс. У самцов же пара половых хромосом образована одной хромосомой Икс, похожей на таковую самок, и другой, более мелкой, названной хромосомой Игрек. Следовательно, женский пол определяется наличием хромосом Икс-Икс, тогда как мужской считается определенным, поскольку его половые хромосомы Икс-Игрек.
Хромосомы Икс являются носителями генов. Напротив, хромосома Игрек играет менее важную роль в наследственности.
Можно легко понять, что все «рецессивные» гены, находящиеся в паре хромосом Икс, проявляются в половой наследственности, потому что у самок их действие часто бывает скрыто доминирующим геном, имеющимся во второй хромосоме Икс. У самцов они иногда выявляются в фенотипе (фенотип — способ проявления генов), так как единственная хромосома Икс, существующая в их генах, лишена гомологической хромосомы, где мог бы находиться доминирующий ген.
Чтобы понять механизм наследственности, точнее передачи информации, нужно иметь в виду, что хромосома Икс является носителем «рецессивного» гена возбудителя болезни.
Предположим, что женщина является носительницей данного гена Икс-Икс и у нее не возникает заболевание, потому что здоровый ген (находящийся во второй Икс хромосоме) доминирует, иными словами, берет верх над больным (находящимся в первой Икс хромосоме). Она выходит замуж за здорового мужчину (Икс-Игрек). Возможные комбинации, которые могут обнаруживаться у детей в результате такого брака, показаны на следующей схеме:
ХХ ХУ ХХ ХУ
Как можно видеть, половина потомства мужского пола будет здоровой (Икс-Игрек), другая половина станет дистрофиками (Икс-Игрек). Тогда как из потомства женского пола половина будет здоровой (Икс-Икс), а другая половина будет являться носителями болезни (Икс-Икс), но ее признаки у них не обнаружатся.
Иначе говоря, в ядре каждой клетки женщины есть две хромосомы Икс, одна дается ей от матери, другая — от отца. В ядре каждой клетки мужчины есть только одна хромосома Икс, которая дается от матери, и маленькая отцовская хромосома Игрек. Здесь-то и заключена сущность проблемы. Дело в том, что в маленькой хромосоме Игрек может находиться лишь ограниченное число генов. Следовательно, в мужской хромосоме Икс содержится наибольшее количество генов, в том числе и «рецессивные» гены. Если в хромосоме Игрек нет алели (пары генов, определяющей характер), которая доминирует и устраняет действие «рецессивной» алели в хромосоме Икс, то создаются условия передачи заболевания.
ХХ ХХ ХУ ХУ
Из приведенных данных становится ясно, почему Дюшенновская мышечная дистрофия (передаваемая «рецессивным» геном, находящимся в одной из материнских хромосом Икс) клинически проявляется только у потомства мужского пола. У женщин не развивается явных симптомов болезни, так как они имеют доминирующий ген в своей второй хромосоме Икс, что обуславливает нормальное развитие у них мышечной системы. Однако женщина является носительницей заболевания. В этом случае практически каждый из ее сыновей будет иметь 50% возможность заболеть дистрофией, тогда как каждая из дочерей будет иметь такую же возможность стать носительницей этого заболевания.
Необходимо также подчеркнуть, что сыновья женщины, носительницы, которым посчастливилось избежать болезни, не будут передавать своим потомкам Дюшенновскую мышечную дистрофию и «рецессивный» ген, вызывающий это заболевание.
2. Некоторые выводы
а) Практически все формы мышечной дистрофии имеют генетическое, или наследственное, происхождение, хотя это не означает, что предки либо потомки больного должны непременно страдать данным недугом. В отдельных случаях, когда болезнь начинается в детстве, она передается внешне здоровыми матерями, а клинически проявляется у детей мужского пола.
b) Но чаще всего потомство больных дистрофией бывает так или иначе подвержено этому заболеванию (у одной его части проявляются клинические симптомы дистрофии, другая же является ее носителем и передает болезнетворные «рецессивные» гены дальнейшим потомкам). Это требует от больных большей ответственности, если они решают иметь детей. На сегодняшний день возможно предложить определенный тест на определение степени наследования Дюшенновской мышечной дистрофии (широко используется с этой целью энзимный анализ). Подобным же способом устанавливают носителя этой болезни. Любой из проконсультированных нами больных согласен с тем, что такой анализ должен стать обязательным для тех, кто собирается вступить в брак или хотел бы это сделать.
с) Когда речь идет о наследственной передаче заболевания, отнюдь не имеется в виду, что предки и ближайшие потомки больного также должны быть дистрофиками. Часто в среднем до и после появления первого больного в семье проходит несколько поколений, у которых нет клинических признаков заболевания.
Приведенные здесь факты не указывают на какое-либо отклонение в половой активности больных мышечной дистрофией. Проблема касается только наследования заболевания их потомством. И в этом смысле нам представляется важным, чтобы больные мышечной дистрофией были детально проинформированы врачами и смогли со всей ответственностью выбирать, иметь или не иметь детей.
Глава 3. СУЩЕСТВУЕТ МНОГО ФОРМ МЫШЕЧНОЙ ДИСТРОФИИ
Как ни покажется странным, сведения об этой болезни очень скудны. Один пациент, которого мы консультировали, говорил об этом так: «Уверяю, что у меня нет никакого представления ни о тех, кто тоже страдают дистрофией мышц, ни о возможностях лечения, ни о связи, которую может иметь заболевание с половой активностью. Я даже не знаю о том, что у дистрофии могут существовать различные формы. Врачи никогда не рассказывали мне обо всем этом подробно, да я и не настаивал, не просил их о такой информации, хотел з н а т ь, н е у з н а в а я. То есть я, конечно, что-то узнавал, но без настойчивости».
Эта нехватка информации о болезни, которая в основе своей является неизлечимой (как правило, удается лишь контролировать и сдерживать ее течение), представляется нам фактом, достойным глубочайшего сожаления.
Огромное разнообразие форм мышечной дистрофии, из которого мы приводим здесь только самые основные, требует сделать простейший вывод: каждый больной имеет право на то, чтобы ему поставили диагноз именно той формы мышечной дистрофии, которой он страдает. Это легко сказать, но гораздо трудней осуществить на практике. Не хотим ли мы невозможного? Ведь обычно считается, сколько людей, столько и мнений. В нашем случае, увы, сколько врачей, столько и диагнозов.
Будет ли хорошо, если каждый больной примется определять свой диагноз, согласно описанию, даваемому нами на последующих страницах? Во-первых, никто не может быть опытным врачом самому себе. Во-вторых, в условиях дефицита информации, как самим больным, так и их близким и друзьям, будет все-таки нелишним обратить внимание на сведения, имеющиеся в этой книге.
1. Псевдогипертрофическая форма (болезнь Дюшенна)
Механизм наследственности обусловлен «рецессивным» геном, зависимым от пола и передаваемым женщиной. Ген вызывает заболевание у потомства мужского пола. У 50% мальчиков могут проявляться клинические симптомы этого заболевания, тогда как 50% девочек могут стать носительницами анормального гена.
Первичные признаки данного заболевания возникают в возрасте от двух до шести лет. Они могут появиться вскоре после рождения ребенка. Диагноз можно установить и до того, как возникнут первые, начальные клинические симптомы, при помощи энзимного анализа крови, биопсии и электромиографии.
Первичные поражения мышц: в начале заболевания поражаются мышцы близкие к области таза, что ведет к нарушениям осанки (лордоз), походка становится качающейся, возникают трудности при спуске и подъеме по лестнице. Через несколько лет также поражаются мышцы плечевого пояса.
Отличительные признаки: мышечное расширение (Псевдогипертрофия) наблюдается в области голени. Оно вызвано большими отложениями жира, которое увеличивается по мере атрофии мышечной ткани.
Прогрессирование болезни протекает быстро, без ремиссий. При этой форме дистрофии оно очень обычно и коварно. Смерть наступает через десять или пятнадцать лет после появления первых клинических симптомов. Однако, применяя антибиотики, такие больные могут прожить значительно дольше. Иногда на последней стадии заболевания у больных отмечаются психические расстройства.
ПРИМЕЧАНИЕ :
В последние годы имеются данные о более благоприятном исходе болезни при некоторых случаях единокровного родства, когда налицо все симптомы, характеризующие Дюшенновскую дистрофию. Подобно описанной выше классической форме, эти случаи напрямую связаны с половой наследственностью и их часто отличает псевдогипертрофия мышц голени. Тем не менее до сих пор невозможно определить, являются ли они разновидностями Дюшенновской дистрофии или представляют собой сходные с ней по симптоматике заболевания.
2. Лицевая, или лопаточно-плечевая форма (болезнь Ландузи-Дежерина)
Механизм ее наследственности объясняется доминирующей автосомой, передаваемой кем-нибудь из предков. Болезнь поражает, как мужчин, так и женщин. Она возникает у 50% детей.
Первичные клинические симптомы, в основном, проявляются в первые годы подросткового периода, а в отдельных случаях с 24 до 26 лет.
Как указывает название болезни, она в первую очередь поражает мускулатуру лица и мышцы плечевого пояса. Неподвижность лица, трудности, испытываемые больным при поднятии рук над головой и при наклонах верхней части туловища вперед, — вот главные признаки, характерные для этой формы мышечной дистрофии.
Прогрессирование болезни, как правило, может идти очень медленно, с относительно долгими периодами задержек. Из всех форм мышечной дистрофии эта, пожалуй, одна из наиболее легких. Продолжительность жизни больного почти не сокращается, хотя в конце концов он становится совершено беспомощным. Как скоро это произойдет, сказать трудно.
3. Лодыжечная форма (включающая ювенильную лопаточную форму, болезнь Эрба)
Механизм наследственности определяется рецессивной хромосомой. Часто аномальный ген присутствует у обоих родителей, и тогда дети не страдают дистрофией. Но когда его имеет только один из родителей или кто-либо из более дальних предков, болезнь проявляется в следующем процентном выражении: у 25% детей будут клинические симптомы болезни, 50% из них будут здоровыми, но носителями аномального гена, и 25% окажутся полностью лишены этого наследственного недуга. Подвержены ему в равной степени, как мужчины, так и женщины.
Первичные признаки заболевания могут отмечаться с конца первого и до третьего десятилетия жизни.
Сначала при этой форме болезни поражаются мышцы близкие к тазовой области и области предплечья. Бывает некоторая слабость икроножных мышц.
Прогрессирование изменчиво. Иногда оно бывает очень замедленным, иногда же, наоборот, наступает быстро, но не настолько быстро, как при Дюшенновской дистрофии. подчас бывает минимальной, и больные могут достигать преклонного возраста.
4. Мышечная дистрофия позднего проявления
Механизм наследственности не установлен. Как известно, данной форме заболевания подвержены и мужчины и женщины. В настоящее время существует мнение, что она не является наследственной.
Первичные клинические признаки болезни возникают на четвертом и пятом десятилетии жизни.
В первую очередь страдают мышцы, прилежащие к области таза.
Прогрессирование также изменчиво. В одних случаях наступает медленно, в других очень быстро.
5. (болезнь Штейнерта)
Механизм наследственности обусловлен доминирующей хромосомой, передающейся кем-либо из предков. Болезнь возникает в равной степени, как у мужчин, так и у женщин. Вероятность случаев такова, что этой формой дистрофии страдает 50% потомства. Однако возможно, что полный синдром не наблюдается у всех членов семьи, пораженной этим заболеванием.
Первичные клинические симптомы обычно появляются в подростковом периоде, хотя симптомы миотонии могут возникать и в зрелости.
При первичном поражении мышц бывает ригидность конечностей, особенно во время нахождения на холоде. Это — один из верных признаков данной формы дистрофии. Также больному бывает трудно разжать сжатые в кулак пальцы.
Прогрессивность болезни Штейнерта такова, что тяжелая инвалидность наступает между 15 и 20 годами после появления первичных клинических симптомов. Мало случаев, когда больные достигают преклонного возраста.
БОЛЕЕ РЕДКИЕ ФОРМЫ МЫШЕЧНОЙ ДИСТРОФИИ
Врожденная мышечная дистрофия. Возникает при рождении. Так же как при неврологическом заболевании, называемом «болезнью Вердинга Хофмана», когда мышцы новорожденного еще так малы, слабы и явно гипотоничны, создаются условия для быстрого прогрессирования дистрофии за довольно короткий срок.
Глазнично-горловая дистрофия. Коварное, быстро прогрессирующее заболевание, которое, как правило, возникает у взрослых. В первую очередь оно поражает наружные глазные мышцы, в болезненный процесс часто вовлекаются и мышцы гортани. Миопатическое выражение лица и особенно непроизвольное опускание верхних век — основные симптомы этой формы, сходные с миостенией.
Дистальная дистрофия. Ее главной особенностью является изначальное поражение основных мышц туловища, которое происходит одновременно с атрофией мышц голени. Это заболевание нервного происхождения. Встречается оно очень редко. Главным образом, им страдают большие семьи. В Швеции число случаев указанного заболевания довольно велико. Причина его по сегодняшний день остается невыясненной.
К врожденным миопатическим дистрофиям относятся и такие заболевания, как митохондрическая , митобулярная и идиопатическая. Их основным признаком является то, что они поражают всех членов семьи и вызывают у них общую мышечную слабость. В большинстве случаев эти формы мышечной дистрофии не прогрессируют.
Глава 4. ГОРАЗДО ВАЖНЕЕ ПРЕДУПРЕДИТЬ…
Профилактика важна при всех болезнях, но крайне необходима в случае мышечной дистрофии. Скольких страданий вы сможете избежать!
Больной мышечной дистрофией, убедившись, что в настоящее время не существует лечения этого заболевания, тотчас настаивает на необходимости раннего обследования и прогноза своего состояния. Ведь это поможет ему спланировать соответствующим образом на будущее свою трудовую деятельность, положение в обществе, досуг и многое другое.
Короче говоря, это означает следующее: когда удается во- время определить, что развивается дистрофия, потенциальный больной может получить профессию с учетом его ограниченных возможностей в будущем, он может завязать дружеские и брачные отношения, которые останутся незыблемыми даже в том случае, если он станет полным инвалидом. Таким образом, больной сумеет не порывать своих связей с обществом, и прогресс заболевания, больший или меньший, не сможет обречь его на удручающее одиночество.
Это означает также, что если нельзя избежать болезни, то возможно хотя бы сдержать ее наступление или замедлить прогрессирование. В то же время больной сможет приспосабливать свою психику к несколько иному ритму жизни, но ему незачем лишать себя доступных удовольствий и быть закомплексованным.
Разумеется, многие больные мышечной дистрофией теперь не получили возможности своевременного обследования. Вызвано ли это нехваткой материальных средств или полным невежеством их самих и их близких, или причина в том, что наши медицинские учреждения очень далеки от того, чтобы предоставлять услуги по раннему прогнозированию течения болезни? Вероятно, тут всего понемногу.
Информация, которую мы предлагаем дальше, открывает, как перед самими больными, так и перед их близкими и всеми гражданами, возможные пути ранней диагностики заболевания, она позволяет настаивать на том, чтобы эти диагностические методы были доступны каждому. Создавайте свои ассоциации либо присоединяйтесь к нам. Пусть будущее не станет кошмаром для последующих поколений больных мышечной дистрофией.
ЧЕМ РАНЬШЕ, ТЕМ ЛУЧШЕ
Разумеется, быстро поставить диагноз можно по первым клиническим симптомам. Но если распад мышечных волокон зашел слишком далеко, наблюдаемые изменения в тканях являются минимальными, что в большой мере затрудняет постановку диагноза.
На начальных стадиях заболевания биохимические и структурные изменения выражены очень ярко и могут определяться, как при помощи электронного микроскопа, так и посредством энзимного анализа. Таким образом удается отличать распад нервных тканей от первичного поражения мышц и вторичной атрофии, а также определять различные формы мышечной дистрофии.
Правильный отличительный диагноз позволяет: а) Применять курс медикаментозного лечения, который соответствует данной форме заболевания. b) Назначать в некоторых случаях комплекс лечебной физкультуры и физиотерапии для поддержания функций здоровых мышц и замедления деструктивного процесса. с) Проводить генетические обследования в семьях, где есть больные, с целью установления при помощи анализов наличия наследственного фактора.
История семьи
Так как все дистрофические заболевания в той или иной степени определяются фактором наследственности, знание о подобных случаях болезни у кого-нибудь из предков помогает установить диагноз.
Биопсия
Биопсия — это изъятие хирургическим путем кусочка ткани пораженной мышцы. Такой кусочек ткани станет химическим препаратом и будет исследован под микроскопом, чтобы установить, какие изменения произошли в мышцах.
Электромиограмма
Электромиограмма — исследование очень похожее на электрокардиграмму, но с той разницей, что для его проведения в мышцы, проверямые на дистрофию, вводятся маленькие электоды в форме иглы.
Анализ на креатин
Когда есть распад мышечной ткани, в крови и моче накапливаются вещества, которые мышцы не могут усваивать. Одним из вернейших показателей подобного нарушения обмена веществ в мышцах является высокое содержание креатина в моче (как мы уже говорили, креатин — это производное аминокислот, которое обычно содержится в мышцах) и низкое содержание креатинина (продукта распада при метаболизме креатина). При дистрофии наблюдается избыточное количество креатина в моче. Однако не усваиваемый креатин также указывает и на многие другие нервномышечные заболевания. Вот почему этот анализ нельзя считать основным в постановке диагноза, а лишь дополнительным, указывающим на то, что есть разрушение мышечной ткани.
Энзимный анализ сыворотки крови
В нормальном состоянии мышцы содержат все клеточные компоненты в мембранах. При дистрофии, когда мышечная ткань разрушается, мембраны клеток становятся чересчур проницаемыми, и вещества, содержащиеся в клетках, фильтруются и поступают в кровь, где их количество может быть гораздо выше нормы. Одним из самых обычных методов установления диагноза служит количественный анализ мышечных энзимов в пробах сыворотки крови. Содержание энзимов в крови повышается сверх нормы в течение начальных стадий дистрофии, задолго до появления первичных клинических симптомов. Но их количество имеет тенденцию к уменьшению по мере того, как обостряется процесс распада мышечных тканей. Возможно, так происходит благодаря немногим сохранным мышцам. Это подчеркивает необходимость ранней постановки диагноза на той стадии болезни, когда биохимическая картина наиболее четкая. Самым важным энзимом в диагностике мышечной дистрофии является креатинофосфокиназа (СК).
Другие энзимы
Также очень важным с точки зрения диагностики может стать повышенное содержание в сыворотке крови таких энзимов, как альдолаза, дегидрогеназа лактика (LDH), трансаминаза глютамика оксалацетика (GOT) и так далее.
Тест на креатинофосфорокиназу (CK)*
Из всех перечисленных форм мышечной дистрофии Аран- Дюшенновская считается самой тяжелой. Поэтому подробнее остановимся на объяснении теста, который, помимо всего прочего, позволяет выявлять женщин- носительниц этой формы дистрофии.
Существует 50% вероятность, что мужское потомство женщины-носительницы будет страдать мышечной дистрофией Дюшенна. Тест на СРК позволяет определять женщин-носительниц. СК — это английская аббревиатура, которой ученые часто пользуются для обозначения энзима «креатин фосфокиназа». (В настоящее время аббревиатура СК употребляется чаще, чем прежняя СРК.)
Энзимами являются вещества протеины, выступающие в роли особых катализаторов. Они находятся во всех клетках и способствуют активизации основных жизненно-важных процессов. Их функции катализаторов ускоряют реакции, без которых невозможны биохимические изменения в клетках и тканях. Таким образом, они могут служить и для определения продолжительности ускоряемых ими реакций. СК участвует в метаболизме, то есть в обмене веществ в мышцах. У человека большие запасы СК находятся в скелетных мышцах.
При мышечных дистрофиях увеличивается проницаемость клеточных мембран в мышцах. В нормальном состоянии клеточные мембраны пропускают продукты распада и задерживают цельные молекулы, как например молекулы энзимов. Когда же мембрана теряет эту избирательную способность, энзимы, которые должны задерживаться, ускользают, и их количество в сыворотке крови значительно возрастает. Высокий уровень содержания в крови энзима СК служит одним из важнейших признаков для постановки диагноза болезни Дюшенна. При других формах мышечной дистрофии содержание энзима СК в сыворотке крови бывает не столь велико (Мышцы, пораженные дистрофией, теряют также и другие энзимы, но ни один из них не является таким важным для диагноза, как СК).
Уровень СК в сыворотке крови также очень высок и у женщин носительниц болезнетворного гена, но не настолько, как у больных дистрофией.
Известно, что существует 50% вероятность того, что мужское потомство женщины-носительницы будет болеть Дюшенновской мышечной дистрофией. Поэтому те женщины, которые знают о том, что в их семьях отмечались случаи такого заболевания, — это могут быть, как сестры и другие близкие родственницы больных, так и более дальние, например, тети и двоюродные сестры по материнской линии, — должны в обязательном порядке пройти тест на СК. Нужно добавить, что этот тест не выявляет носительниц других форм мышечной дистрофии.
Тест безболезнен. Он состоит в том, что из вены на руке женщины с подозрением на болезнетворный ген берут небольшую пробу крови (5 c.cm. или меньше). Ее сыворотку используют для определения путем клинического анализа уровня содержания энзима СК. Эффективность теста составляет приблизительно от 70 до 80% .
У женщин носительниц дистрофии Дюшенна, как правило, не возникает никаких клинических признаков этого заболевания, хотя в некоторых случаях могут наблюдаться такие симптомы, как небольшое увеличение мышечной массы, и возможна до определенной степени слабость мышц, которая никогда не прогрессирует.
Конечно, у носительниц Дюшенновской болезни проявляется определенная мышечная патология. Если исследовать пробу их мышечной ткани под электронным микроскопом, то можно обнаружить аномальную разрозненность мышечных волокон наподобие той, которая встречается у мужчин с этой болезнью в доклинической стадии. Женщины, имеющие такую мышечную аномалию, страдают так называемой субклинической формой мышечной дистрофии, которая не прогрессирует. Должно быть поражено 50% мышечной ткани прежде, чем мышцы потеряют положенные им функции.
Эффективность теста больше у молодых женщин, чем у находящихся в зрелом возрасте, поскольку уровень СК с возрастом понижается, как у больных, так и у носителей дистрофии Дюшенна. Нужно добавить в заключение, что тест с отрицательным показателем не исключает полностью вероятность того, что женщина может быть носителем болезни.
Возвращаемся к тому, с чего начали
Теперь вернемся к началу этой главы, чтобы еще раз напомнить: мышечную дистрофию необходимо предупреждать. Своевременный диагноз не устранит болезнь, но в значительной степени уменьшит ее последствия.
Давайте же используем все средства, имеющиеся в наших руках, чтобы избегнуть ненужных страданий самим и помочь избавиться от них последующим поколениям. Все мы имеем право на лучшее будущее.
Глава 5. НО НУЖНО ТАКЖЕ ЛЕЧИТЬ (ИЛИ ПОМОГАТЬ БОЛЬНЫМ)
Все, что мы сказали о ранней диагностике заболевания, возможно, прозвучало для многих больных дистрофией райской музыкой. Вот если бы им в свое время поставили верный диагноз! И это вполне справедливо.
Необходимо четко определить, что можно сделать для того, чтобы хоть как-то облегчить жизнь тех, кто сегодня в большей или меншей степени страдает мышечной дистрофией. И как бы ни было тяжело, но нужно признать, что на сегодняшний день это заболевание является неизлечимым и в той или иной мере прогрессирующим. Очень часто здоровый, но обладающий малой культурой человек психлогически ранит больного примерно таким вопросом: «Как!? Ты же раньше мог все делать сам?» Ответ последует незамедлительно: «Ясно, дружище, ведь э т о прогрессирует».
Психологическая реабилитация
Письма, которые нам прислали некоторые из консультируемых нами больных, содержат много интересных суждений. Эти письма и легли в основу данного подзаголовка. Иногда их писали сами больные, иногда их бизкие и друзья. С большим удовольствием мы помещаем здесь выдержки из их писем.
«Нужно, чтобы больные как можно раньше адаптировались к новой ситуации и располагали средствами, облегчающими их социальную интеграцию…» Это нелегкая задача, но решение ее возможно и нужно в качестве отправного пункта.
Единодушно мнение о том, что необходимо избавляться от излишней опеки родных и друзей. «Пусть близкие ограничат свою опеку тем, в чем больной не может обойтись без их помощи, а во всех других аспектах воспринимают его как полноправного члена семьи»… «пусть нас перестанут постоянно опекать и дадут нам возможность делать то, что мы еще в состоянии делать самостоятельно»… и так далее и тому подобное. Никакая, даже самая преданная любовь не может служить оправданием для чрезмерной опеки.
Преодоление комплексов, желание надеяться на лучшее, мысли о том, как суметь по возможности полнее участвовать в жизни — вот о чем в той или иной форме нам пишут все консультируемые у нас пациенты, ставшие нашими друзьями. «Первое, что я бы посоветовал своему товарищу по несчастью, также как я страдающему от мышечной дистрофии, — это победить комплекс неполноценности в том, что в физическом отношении ты не такой, как все, — если он у него имеется. И второе — это принимать большее участие в жизни, например, начать учиться, вступить в спортивный или любой другой клуб…» «Не дать болезни заранее сломить себя, искать возможности компенсации физической немощи, развивая интеллектуальные способности участием в культурных мероприятиях, занятиями умственным трудом. Потому что мозг ни в какой степени не бывает затронут болезнью…» Это настолько очевидно, что не нуждается в комментариях с нашей стороны.
Кажущейся безнадежностью, только кажущейся проникнут призыв жить каждым днем: «…Я сказал бы такому же больному, чтобы он «цеплялся» за каждую минуту и, убедившись в том, что дальше будет хуже, — радовался, грустил и работал каждую минуту так, словно она последняя, и не думал о следующей».
Надежда для большинства наших пациентов и их близких стала неотъемлимой частью жизни. Они пишут нам: «Находясь целыми днями в корсете и ортопедических аппаратах, мы не унываем, хотя бы потому, что видим солнце и можем радоваться жизни…»
Физическая реабилитация
Больные дистрофией мышц хорошо знают, что от реабилитации они не могут ожидать чудес. Реабилитация — это средство поддержания себя в относительной форме, тормоз, сдерживающий прогрессирование болезни. Им известно также и то, что существует первая реабилитация, которая зависит от них самих. И они учат этому своих товарищей. «…Пусть он делает всевозможные физические упражнения и не принимает никакой посторонней помощи в тех действиях, которые ему доступны, чтобы дистрофия прогрессировала как можно медленнее…» «… Больные должны делать все, что могут, и двигаться, как могут, сами. Пусть они поменьше лежат в кровати…»
Необходима также физическая реабилитация в медицинских учреждениях. Рассуждать об этом, подчеркивая важность такой реабилитации, можно бесконечно. Но как осуществить ее на практике?
Один из наших корреспондентов настаивает на определении «адекватная реабилитация», которое заслуживает отдельной темы: «Адекватная реабилитация, я говорю, «адекватная» потому, что избыток физических упражнений, несоответствующий возможностям больного, может принести гораздо больше вреда, чем отсутствие всякой физической реабилитации. Поэтому крайне необходимо, чтобы физиотерапевт глубоко изучил специфические особенности этого заболевания…»
Действительно, реабилитация больного мышечной дистрофией существенно отличается от реабилитации человека, страдающего полиомиелитом. К глубочайшему сожалению, не все врачи понимают это.
В заключение нам хочется поблагодарить Американскую Ассоциацию Прогрессирующей Мышечной Дистрофии, информативные буклеты которой очень помогли нам в работе над этой брошюрой. Мы сочли возможным привести здесь ее адрес, чтобы заинтересованные читатели смогли получить необходимые им сведения: MUSCULAR DYSTROPHY ASSOCIATION.810 Sevenh Avenue, New York, N.Y. 10019. USA.
Перевод с испанского В.М. Труфанова