Neuromuskulaarsete haiguste ravi lastel. pärilikud lihashaigused
Närvisüsteemi progresseeruvad degeneratiivsed haigused tekivad geneetiliselt määratud patoloogia või embrüonaalse arengu defekti tagajärjel. Nende haiguste tavalised ilmingud on: närvikoe degeneratiivne iseloom ja süsteemsed kahjustused, progresseeruv kulg. Nende hulka kuuluvad eelkõige süringomüelia, mille puhul tekivad seljaajus pikad õõnsused, mis hävitavad tagumisi sarvi. See toob kaasa valu- ja temperatuuritundlikkuse defekti, atropaatia.
Pärilike ataksiate rühm on üsna arvukas, nende peamine ilming on ataksia, mis on seotud väikeaju radade patoloogiaga või sügava tundlikkusega.
Amüotroofiline lateraalskleroos (ALS)- kõige raskem ja kiiremini progresseeruv seljaaju kanalite haigus. Sel juhul tekib atroofilise pareesi ja püramiidsümptomite kombinatsioon, see tähendab, et arenevad motoorsed häired on nii perifeerse kui ka tsentraalse halvatuse iseloomuga.
Parkinsoni tõbi on progresseeruv haigus, mis põhineb mustaine tiheda osa pigmenti sisaldavate dopamiinergiliste neuronite ja tüve teiste pigmenti sisaldavate tuumade esmasel kahjustusel. Patsiendi lähisugulaste risk on ligikaudu 10 korda suurem kui üldpopulatsioonis. Parkinsoni tõbe iseloomustab sümptomite kolmik: treemor, suurenenud lihastoonus ja hüpokineesia; Diagnoosimise kriteeriumid on väga keerulised. Haiguse kliinik ilmneb ainult siis, kui enam kui 80% neuronitest sureb. Mõnel juhul debüteerib haigus enne 18. eluaastat (nn juveniilne parkinsonism), enamasti vanemas eas. See seab suuremad nõudmised ravi õigeaegsusele ja piisavusele.
Kaasaegne adekvaatne teraapia pakub palju võimalusi kahjustatud funktsioonide heaks kompenseerimiseks, samuti sotsiaalse kohanemise säilitamiseks või taastamiseks.
Pärilikud neuromuskulaarsed haigused
Degeneratiivsed neuromuskulaarsed haigused- päriliku iseloomuga neuromuskulaarse aparaadi valdava kahjustusega haigused. Neid nimetatakse ka progresseeruvateks neuromuskulaarseteks düstroofiateks (PMD) ja need moodustavad kõigi pärilike haiguste suurima rühma.
Progresseeruvate lihasdüstroofiate klassifikatsioon
- Progresseeruvad lihasdüstroofiad on geneetiliselt määratud häired, millega kaasnevad esmased progresseeruvad degeneratiivsed muutused lihastes (ilma perifeerse motoorse neuroni primaarse patoloogiata). Nende puhul on just lihaskoe esmase geenidefekti sihtmärk, mille tõttu toimub lihasvalgu müodüstrofiini ebanormaalne süntees ja selle lagunemine kiireneb. Närvisüsteemi kahjustus müopaatia korral on sekundaarne.
- Spinaalne amüotroofia on seljaaju eesmiste sarvede esmane geneetiliselt määratud kahjustus, millega kaasneb sekundaarne progresseeruv perifeerne halvatus ja lihaste atroofia.
- Neuraalne amüotroofia - polüneuropaatia esmane geneetiliselt määratud sündroom (müelinopaatia tagajärjel) koos sekundaarse amüotroofia ja vegetatiivse-sensoorsete häirete tekkega.
Primaarsed progresseeruvad lihasdüstroofiad
Erinevad haigusvormid ilmnevad erinevas vanuses - alates 1-2 aastast kuni 40-50 ja vanemad. Neid iseloomustab motoorne kohmakus, ebastabiilsus, kukkumised kõndimisel, väsimus. Lapsel on hirm ja soovimatus kõndida. Moodustatud kõnnakuga patsientidel esineb "pardi kõnnak" - kahlamine.
Mõnele vormile on iseloomulik lihaste pseudohüpertroofia, sagedamini haigestuvad säärelihased: nende atroofia koos atroofia varjamisega ja isegi suuruse suurenemine side- ja rasvkoe kasvu tõttu. Nõrkus ja lihaste atroofia lokaliseeritakse esialgu vaagnavöötme lihastes, maksimaalne raskusaste on proksimaalsetes jalgades.
On väljendunud nimmepiirkonna lordoos, skolioos, "pterygoid" abaluud, kitsas "herilase" vöökoht. Istumisasendist tõusmine on keeruline ja lapsed kasutavad abitehnikaid (Gowersi tehnikad) - “redeliga ronimine”, “ise ronimine”. On olnud dementsuse juhtumeid. Südamelihas kannatab. Lisaks kaotavad patsiendid võime iseseisvalt kõndida. Protsessi on kaasatud kardiovaskulaarsüsteem (tekib laienenud või hüpertroofiline kardiomüopaatia).
Sekundaarne - seljaaju ja neuraalsed lihasdüstroofiad
Lülisamba lihasdüstroofiad (amüotroofiad) päritakse autosoomselt retsessiivselt. Spinaallihaste atroofia geen on kaardistatud kromosoomil 5gl1.2-13.3.
Tabelihäirete varajasi märke võib tuvastada. Motoorse arengu hilinemine. Elektromüograafia läbiviimisel tuvastatakse seljaaju eesmiste sarvede kahjustus. Haiguse kulg on progresseeruv.
Amüotroofia seljaaju vormide korral registreeritakse virvenduspotentsiaalid puhkeolekus elektromüogrammil; impulsi levimise kiirus piki jäsemete närve on suhteliselt terve, kuid võib väheneda ka seljaaju motoorsete neuronite surma tagajärjel.
Neuraalse amüotroofia kõige levinum variant on Charcot-Marie-Toothi neuraalne amüotroofia. Kliiniliste ilmingute poolest sarnaneb see sensomotoorsele polüneuropaatiale, distaalselt rõhutatud ja algab jalalabadest ja säärtest. Voolab hästi, aeglaselt. Aja jooksul moodustub jalgadele iseloomulik deformatsioon - vastavalt "toonekure jalgade" või "ratsutamispükste" tüübile: tervete reielihastega sääre atroofia tagajärjel õhukesed. Esiteks "kukkuvad välja" Achilleuse refleksid, seejärel vähenevad põlverefleksid.
Elektroneuromüograafiaga registreeritakse jäsemete närvide kaudu impulsi levimise kiiruse järsk vähenemine.
Müasteenia. Müasteenilised ja kolinergilised kriisid
myasthenia gravis(miastenia gravis pseudoparalitica) on raske autoimmuunse iseloomuga neuromuskulaarne haigus, mida iseloomustab vöötlihaste patoloogiline väsimus ja nõrkus (Akimov G. A., Odinak M. M., 2000).
Etiopatogenees. Peamine seos on autoantikehade tekkimine lihaskiu otsaplaadi nikotiini kolinergiliste retseptorite vastu ja neuromuskulaarse ülekande blokaad. Myasthenia gravise patogeneesi ja prillinäärme harude kahjustuse vahel on seos. Sageli tuvastatakse tümoom (kuni 40% juhtudest), harvem - harknääre atroofia.
Kliinik. Myasthenia gravis võib tekkida igas vanuses, kuid sagedamini vanuses 16–40 aastat, kuid esineb nii varasemaid kui ka hilisemaid vorme (haigestumise tipp on 30–70 aasta vanuses). Naised haigestuvad sagedamini kui mehed. Peamine sümptom on lihaste patoloogiline väsimus koos nende suureneva nõrkusega korduvate liigutuste ajal, näiteks lugemisel kahekordistumine või ptoos.
Myasthenia gravis'ega tekkiv lihasnõrkus erineb perifeersest või tsentraalsest pareesist selle poolest, et liigutuste kordumisel, eriti kiires tempos, suureneb see järsult ja võib ulatuda täieliku halvatuseni. Pärast puhkust, und võivad esimesed liigutused olla normaalsed, kuid ilmneb järgnev väsimus, mille määr koormuse jätkumisel edeneb.
Müasteenia episood võib areneda vastsündinutel, kelle emad on sündinud müasteeniaga (nn vastsündinute myasthenia gravis). Liikumishäirete hüvitamise määr võib olla täielik, piisav (koduseks iseteeninduseks), kehv (vajab välist abi). Myasthenia gravise kõige hirmutavam tüsistus on müasteeniline kriis.
müasteeniline kriis- äkiliselt tekkiv kriitiline hädaolukord neuromuskulaarse juhtivuse blokeerimise tagajärjel. Peamised sümptomid on kiiresti arenev üldine lihasnõrkus, mis ulatub tetrapleegia astmeni.
Tüsistused:
- bulbarvormiga hingamispuudulikkus,
- hingamisteede obstruktsiooni oht paksu röga kogunemise tõttu,
- toidu aspiratsiooni või "klapi lämbumise" võimalus keele tagasitõmbumise ja epiglottise nõrkuse tõttu;
- diafragma rike ja roietevaheliste hingamislihaste nõrkus.
Antikoliinesteraasi ravimite üleannustamine võib viia kolinergilise kriisi tekkeni koos heaolu järsu halvenemisega. Vältimatu abi” osutavad intensiivravi osakonnas või intensiivravi osakonnas (palatis) asuvad meditsiinitöötajad.
Iv. Vaptsarov
Lihashaigused on lapsepõlves suhteliselt tavalised. Mõned neist on tingitud lihaskiu esmasest kahjustusest. Need on kaasasündinud, geneetiliselt sõltuvad (pärilikud ja pärilikud-perekondlikud) haigused. Teised kujutavad endast metaboolsete häirete, nakkuslike, põletikuliste ja toksiliste protsesside tagajärjel tekkinud lihaskahjustusi. Kolmanda rühma haigused on põhjustatud närvisüsteemi ja neuromuskulaarse aparatuuri haigustest. Samuti on rühm, mis ühendab seni selgitamata etioloogiaga lihashaigusi.
ESMASED JA PÄRILIKUD LIHASHAIGUSED
PROGRESSIIVNE LIHASDÜSTROOFIA
Progresseeruvad lihasdüstroofiad on geneetiliselt määratud pärilikud ja pärilikud perekondlikud haigused, mida iseloomustab krooniline progresseeruv areng, mille tagajärjeks on raske puue. Nende haiguste suhteline esinemissagedus, mis viimasel ajal on suurenenud, kliiniliste sümptomite raskus, samuti spetsiifilise ja tõhusa ravi puudumine muudavad need sotsiaalseteks haigusteks.
Patogenees ja patoloogiline anatoomia. Primaarsete lihasdüstroofiate histoloogilist pilti iseloomustab ebaühtlane segmentaalne degeneratsioon, mis areneb piirkondades piki lihaskiudusid, mis nendes piirkondades kaotavad oma põikiriba. Sarcolemma tuumade suurus suureneb, need muutuvad ümaramaks ja asuvad keskpunktile lähemal. On pilt hüaliinsest, granulaarsest või vakuolaarsest düstroofiast, millel on iseloomulik kalduvus teatud määrdumisele. Nähtavad fagotsütoos, sidekoe vohamine ja oluline rasvatilkade kogunemine kiudude vahele, mis annab düstroofselt väljendunud lihasele kollaka värvuse. Eriti iseloomulik tunnus on aga kahjustuste juhuslik jaotus üksikutes kimpudes ja seetõttu on nende suurused erinevad. See omadus eristab progresseeruvaid lihasdüstroofiat närvilistest, kus eesmiste sarvede, juurte või tüve kahjustus põhjustab lihaskiudude süstemaatilist ja ühtlast, mitte segmentaalset atroofiat.
Nende düstroofiate patogeneetilisi mehhanisme ei ole veel täielikult välja selgitatud. Praegu on kõige vastuvõetavam ensüümiteooria, mille kohaselt tekivad lihaskiu düstroofsed muutused lihaste aldolaasi, fosfokreatiinkinaasi ja vähemal määral ka laktaatdehüdrogenaasi aktiivsuse rikkumise tagajärjel. Haiguse esimestel faasidel nende ensüümide tase veres tõuseb, kuid paralleelselt atroofia progresseeruva arenguga väheneb neid tootva aktiivse lihaskoe vähenemise tõttu järk-järgult. Transaminaaside tase on tavaliselt normaalne. Hüperkreatinuuria ja hüpokreatinuuria tuvastatakse ka madala kreatineemiaga.
Elektromüogrammi iseloomustavad: a) elektrilise aktiivsuse puudumine puhkeolekus; b) mootoriüksuste madalad, kõverad ja mõnikord mitmefaasilised potentsiaalid; c) suureneva pingutusega häirekõverate kiire tekkimine; d) maksimaalse kontraktsiooni korral häirete salvestamise taustal kontrasteerub väljendunud lihasnõrkus.
Sõltuvalt haiguse geneetilise ülekande tüübist ja protsessi esialgsest lokaliseerimisest teatud lihasrühmades on üldiselt aktsepteeritud, et progresseeruvad lihasdüstroofiad esindavad mitmeid kliinilisi ja geneetilisi vorme.
Duchenne'i haigus(paralysis pseudohypertrophicans, paralysis myosclerotica) on X-seotud retsessiivne haigus, mis avaldub ligikaudu aasta pärast sündi ja peamiselt poistel. Seda iseloomustab kehatüve lihaste ja jäsemete proksimaalsete osade tugevuse üldine ja progresseeruv vähenemine ning jäsemete distaalsete osade lihaste suhteliselt säilinud motoorne jõud. Kõigepealt on kahjustatud alajäsemete lihased. Kõnn omandab “pardi” iseloomu.Kõndimisel jääb keha maha kiiresti areneva lordoosi tõttu.Lapsed kukuvad ja ronivad trepist sageli vaevaliselt.Püüdes üles tõusta rullub ümber kõhuli, toetub omale. käed, painutab aeglaselt põlvi ja alles pärast seda ajab end kätega aidates sirgu, nagu eespool kirjeldatud."Siis katab kahjustus ülemiste jäsemete proksimaalseid lihaseid ja õlavöötme. Kaugelearenenud juhtudel abaluu atroofia lihased viib mõnikord abaluu alatae ilmumiseni.Pärast suuri lihaseid mõjutavad väiksemad lihased.Jäsemete kõikide lihasrühmade sümmeetrilise, kuid ebaühtlase kaasatuse tõttu on lülisamba tugevad deformatsioonid ja kõverus.Nägu ei ole tavaliselt m abiellub. Mõnedes suuremates lihastes toimub koos kiudude atroofiaga sidekoe vohamine ja rasvade kogunemine, mille tulemuseks on pseudohüpertroofia, mis on Duchenne'i tõve iseloomulik tunnus. See protsess väljendub kõige selgemalt nelipealihastes, harvemini deltalihastes, mille mass on kontrastiks naaberlihaste atroofia taustal.
Reeglina jäävad kõõluste refleksid normaalseks, kuid tegelik lihaskontraktsioon nõrgeneb järsult.
Düstroofiline protsess võib mõjutada ka müokardit. Valkude ja rasvade degeneratsiooni ning fibroosi tagajärjel tekib kardiomegaalia. EKG näitab PQ laienemist, sageli ühe jala blokaadi ja T segmendi vähenemist. Pulss kiireneb ja lõppstaadiumis ilmnevad kardiovaskulaarse nõrkuse sümptomid. Atroofia ja liikumispiirangu tõttu täheldatakse osteoporoosi, „diafüüsi hõrenemist ja harvadel juhtudel luumurdu.Progresseeruv puue võib põhjustada muutusi iseloomus, kuid vaimse arengu mahajäämust täheldatakse harva.
Leideni haigus - Möbius on Duchenne'i tõve tüüp, mida iseloomustab pseudohüpertroofia puudumine ja protsessi lokaliseerimine ainult vaagna ja alajäsemete lihastes. See on pärilik - autosoomne retsessiivne tüüp.
Landouzy haigus - Dejerine nimetatakse Myopathia facio-scapulo-humeralis, kuna protsess algab näolihastest ja mõjutab valdavalt õlavöötme lihaseid. See pärineb autosomaalselt domineerival viisil ja mõjutab mõlemat sugupoolt võrdselt. Tavaliselt avaldub see teisel elukümnendil, kuid on kirjeldatud ka varasema ja hilisema alguse juhtumeid. Näolihaste düstroofia tulemuseks on iseloomulik müopaatiline nägu, millel on fikseeritud ilme, horisontaalne naeratus ja silmade mittetäielik sulgemine une ajal. Atroofia katab järk-järgult õlavöötme lihased (mm. dentatus, rhomboideus, trapezius, infra- et supraspinosus, m. m. pectorales, deltoideus, biceps et triceps brachiaiis jt), põhjustades liigutuste ja õla pterigoidse kuju olulise piirangu. labad (scapulae ala-tae) . Iseloomulik on pseudohüpertroofia puudumine enamikul patsientidel. Mõjutatud on ka müokard, kuid tavaliselt puuduvad kliinilised sümptomid ja diagnoos tehakse EKG abil. See haigusvorm areneb palju aeglasemalt, eksisteerib aastaid. Tema prognoos on hoolimata progresseeruvast puudest suhteliselt parem.
Erbi tõbi (Myopathia scapulo-humeralis) edastatakse autosoomselt domineerival viisil. Kliiniliste omaduste ja arengu poolest on see väga sarnane Landouzy-Dejerine'i haigusele, kuid erineb sellest näolihaste puudumise või hilise kahjustuse ning pseudohüpertroofia esinemise poolest.
Haruldased histoloogilised sordid
Duchenne'i tõve vastsündinu vorm sarnaneb kliiniliselt täielikult Oppenheimi tõvega (sündroom, mis varem ühendas nii primaarset kui ka neuraalset lihasdüstroofiat, vt Werdnig-Hoffmanni tõbi).
Primaarne kaasasündinud generaliseerunud Krabbe lihaste hüpoplaasia ja sellega seotud Batten-Turneni tõbi.
Keskne tuumhaigusmida iseloomustab müofibrillide rühmitus kimbu keskel ja ketta puudumine. Kliiniline pilt koosneb mittearenevast müotooniast, mis hiljem areneb selgelt väljendunud lihasnõrkuseni. See edastatakse autosomaalsel domineerival viisil.
Nemaliini müopaatiaon sarnane kliiniline pilt, kuid histoloogilisel uuringul leitakse Z-ketas, mille tropomüosiin moodustab sarkolemma all spetsiaalsed "pulgad".
Müotubulaarsed müopaatiad histoloogiliselt koosnevad nad loote tüüpi lihastest, mille kiu keskel on toruõõnsused, mis sisaldavad suurt hulka mitokondreid.
Mitokondriaalsed müopaatiad eristuvad mitmesugused mitokondriaalsed anomaaliad: kandmised, hiiglaslik suurus või ebatavaliselt suur arv. Pärandi tüüp on autosoomne dominantne.
Progresseeruva lihasdüstroofia diagnoos üksikasjaliku kliinilise pildi juuresolekul on lihtne teha isegi esimesel läbivaatusel. Klassikaliste vormide eristamine on võimalik kahjustuse esialgse rühma, pseudohüpertroofia olemasolu või puudumise ja anomaalia edasikandumise geneetilise tüübi kindlaksmääramisel.
Haiguse arengu varases staadiumis, aga ka ebatüüpiliste, kustutatud vormide korral tuleb silmitsi seista suurte raskustega. Nendel juhtudel aitavad täpset diagnoosi panna perekonna geneetilised ja biokeemilised (ensüümi) uuringud.
Kogu rühma diferentsiaaldiagnostikas on vaja arvestada neuraalsete (Werdnig-Hoffmann, Kugelberg-Wellander) ja teiste sümptomaatiliste lihasdüstroofiate, müatoonia ja müotooniaga varajases eas jne.
Kliinik ja prognoos. Lihaste tagasitõmbumine ja kõõluste atroofia (nende vormide sümptomid) viivad järk-järgult kontraktuuride ja liigeste deformatsioonide tekkeni, mis kahjustavad lapse motoorseid funktsioone ja liigutusi. Teisest küljest kiirendab tegevusetus atroofiat, tekitades nõiaringi, mis lõpeb täieliku invaliidistusega. Duchenne'i, Leideni - Möbiuse ja Landouzy - Dejerine'i vormide prognoos halveneb progresseeruva müokardi düstroofia ja nende laste kalduvuse tõttu hingamisteede infektsioonidele. Progresseeruv puue mõjutab negatiivselt laste psüühikat, samas kui raskemate vormidega võib kaasneda teatav neuropsüühilise arengu viivitus. Sagedamini täheldatakse patoloogilisi muutusi iseloomus.
Narkootikumide ravi (adrenaliin, pilokarpiin, ezeriin, galaktamiin, nivaliin, proteolüsaadid, androgeensed anaboolsed hormoonid, E-vitamiin, glükokool, isegi adenosiintrifosforhape) ei anna märkimisväärseid tulemusi. Suuremal määral võite loota füsioteraapiale, mis parandab lihaste vereringet: soojad protseduurid, kerge massaaž jne. Samuti on ette nähtud vasodilataatorid, näiteks vasculat.
Täielik puhkus peegeldub ebasoodsalt. Laps peab tegema mõõdukaid, doseeritud aeglases rütmis liigutusi, mis ei põhjusta lihaste energiavarude ammendumist ega põhjusta seisundi halvenemist. Õige psühhopedagoogiline lähenemine on eriti vajalik oma haigust sügavalt põdevate laste meeleolu parandamiseks.
NÄRVISÜSTEEMI KAHJUSTUSEST PÕHJUSTATUD PÄRILIKU LIHASATROOFIA
Neuraalne lihasatroofia (Charcot-Marie-Toothi tõbi) - perifeerse närvisüsteemi pärilik degeneratiivne haigus.
Spinaalne lihaste atroofia (Werdnig-Godfmanni tõbi). Selle haiguse peamine patoloogiline protsess on seljaaju eesmiste sarvede motoorsete rakkude progresseeruv degeneratsioon. Lihaste atroofia on sekundaarne nähtus.
Haiguse etioloogiat ei ole täielikult välja selgitatud.
Patoloogiline anatoomiline uuring tuvastab seljaaju eesmiste sarvede rakkude kahjustuse.
Kliinik. Haigus avaldub esimestel elupäevadel või esimestel kuudel. Tekib ebatavaliselt raske lihaste hüpotensioon, mis algab proksimaalsetest alajäsemetest ja levib kiiresti kogu skeletilihastesse. Laps lamab täiesti loidult, ilma toonuseta ja teeb väikseimate liigestega (näiteks sõrmedega) vaid kergeid liigutusi. Kuid näoilmete elavus vastandub teravalt jäsemete üldise loiduse ja lapse vaikse nõrga häälega. Passiivsed liigutused on võimalikud igas suunas ja liigesed jätavad mulje erakordsest lõtvusest. Hüpotensioon mõjutab järsult abihingamislihaseid, mistõttu hingamine ja kopsuventilatsioon on väga rasked. Sellest tuleneb atelektaatilise kopsupõletiku eriline sagedus ja hingamisteede infektsioonide tõsine kulg. Lihaste atroofia on väga väljendunud, kuid pilti varjab märkimisväärne nahaalune rasvkude. Röntgenpiltidel on aga lihaste hõrenemine selgelt näha. Pareesi ja halvatuse esinemine väljendub kõõluste reflekside nõrgenemises või täielikus puudumises olemasolevate nahareflekside taustal, samuti lihaste tegelikus kontraktsioonis. Elektrilise erutuvuse uurimine paljastab kronaksia pikenemise ja lihaste degeneratsiooni reaktsiooni ning elektromüogramm neurogeense lihasatroofia.
Sellel haigusel on autosoomne retsessiivne ülekandemuster. See on tavaks jagada kolmeks peamiseks kliiniliseks vormiks: varajane (kaasasündinud), lapseea ja hiline (Kiegelberg-Welanderi tõbi). Viimasel ajal on kirjeldatud ka vahevorme.
Lülisamba lihaste atroofia varajane vorm avaldub juba emakasse loote puudumise või täiesti loid liigutustena, mis teeb muret eelkõige nendel rasedatel, kes on juba terve lapse sünnitanud.
Diagnoos tehakse kohe pärast sündi, kuna see jätab mulje teravast hüpotensioonist ja lapse liikuvuse vähenemisest. Tulevikus süvenevad hüpotensioon ja parees jätkuvalt. Lapse nägu kaotab täielikult oma näoilmed.
See haigusvorm langeb täielikult kokku Oppenheimi kirjeldatud kaasasündinud müotooniaga, mida ei ole hiljuti peetud iseseisvaks haiguseks, kuna nende haiguste üldised sümptomid võimaldasid ühendada need üheks nosoloogiliseks üksuseks.
Varaste vormide prognoos on raske. Lapsed surevad imikueas hingamisteede infektsioonidesse. Hiliste ja kergete vormide korral, kui lapsed ei sure esimese kolme eluaasta jooksul, võib toimuda märkimisväärne kohanemine.
Ravi. Soovitatav on kasutada kõiki poliomüeliidi raviaineid. Kõige olulisem on aga hingamisteede infektsioonide ja nakkushaiguste ennetamine, millesse need lapsed tavaliselt surevad.
Clinical Pediatrics Toimetanud prof. Br. Bratinova
Lk 40/44
NEURO-LIHASHÄIRETE KLASSIFIKATSIOON
Perifeerse närvisüsteemi sensoorseid ja motoorseid häireid nimetatakse tavaliselt neuromuskulaarseteks haigusteks. Tavaliselt osalevad nad ühe või enama seljaaju reflekskaare elemendi protsessis: seljaaju eesmiste sarvede rakud, motoorsed närvikiud, neuromuskulaarsed sünapsid, lihased ja lihaseid ja kõõluseid innerveerivad sensoorsed närvikiud (joonis 21-1). ). Selle reflekskaare elementide kahjustus põhjustab kõõluste reflekside pärssimist, mis on iseloomulik kõigile neuromuskulaarsetele haigustele. Lisaks täheldatakse tavaliselt lihaste nõrkust ja atroofiat.
Klassifikatsioon
- Seljaaju eesmiste sarvede rakkude kahjustus Werdnig-Hoffmanni tõbi
Lastehalvatus
Muud viirusinfektsioonid
- Polüneuropaatiad
Nakkusjärgne polüneuriit (Guillain-Barré sündroom)
Difteeria polüneuriit
Toksilised neuropaatiad (raskmetallimürgitus), ravimitest põhjustatud neuropaatiad, metaboolne polüneuropaatia (vt tabel 21-2) hüpertroofiline interstitsiaalne neuriit (Dejerine-Sotti tõbi) Charcot-Marie-Toothi haigus (peroneaalne lihasatroofia) Kaasasündinud sensoorne neuropaatia Valu kaasasündinud sensitiivsus
- Mononeuropaatiad Kaasasündinud ptoos
okulomotoorse närvi halvatus (Tholosa-Hunti sündroom)
VI paari kraniaalnärvide halvatus (Duane'i sündroom)
Näo halvatus (Belli halvatus)
Erbi halvatus Peroneaalne halvatus Istmikunärvi vigastus
- Neuromuskulaarsete sünapside haigused Myastenia gravis
Botulism
- Lihashaigused Põletikulised protsessid Polümüosiit
Müosiit ossificans Endokriinsed või metaboolsed müopaatiad Müopaatia hüpertüreoidismi korral
Riis. 21-1. Neuromuskulaarse süsteemi moodustavate struktuuride skemaatiline esitus.
1 - eesmise sarve rakk; 2 - motoorne närvikiud; 3 - motoorne närvilõpp lihases; 4 - lihas; 5 - sensoorne retseptor lihases (lihase spindel) - 6 - sensoorne närvikiud.
Müopaatia hüpotüreoidismi korral
Kortikosteroidravist tingitud müopaatia
Lihaskarnitiini puudus
Üldine karnitiini puudus
Kaasasündinud lihasdefektid
Lihaste puudumine
Kaasasündinud tortikollis
Kaasasündinud müopaatiad (keskse südamiku haigus ja mittekarmiinpunane müopaatia)
Mitokondriaalne müopaatia
Kaasasündinud müotoonia (Thomseni tõbi)
Perioodiline halvatus
Hüperkaleemiline vorm (adynamia episodica hereditaria) Hüpokaleemiline vorm Paroksüsmaalne müoglobinuuria karnitiinpalmitüültransferaasi puudulikkus McArdle'i tõbi lihasdüstroofia
Pseudohüpertroofiline vorm (Duchenne)
Kaasasündinud lihasdüstroofia
SELJAAJU EESMISE SARVEDE RAKU KAHJUSTUS
Seljaaju eesmiste sarvede rakkude selektiivne kahjustus esineb lastehalvatuse ja mõnikord ka teiste viirusnakkuste korral, sealhulgas Coxsackie ja ECHO viiruste põhjustatud infektsioonide korral. Nende pärilik degeneratsioon avaldub peamiselt imikueas.

Riis. 21-2. Lihaskoe fassikulaarne atroofia (a), eesmiste juurte kahvatus (b) ja motoorsete neuronite degeneratsioon (c) Werdnig-Hoffmanni tõve korral.
Varases eas spinaalne lihasatroofia. Werdnig-Hoffmanni tõbi pärineb retsessiivselt. Esmane patoloogiline tunnus on seljaaju eesmiste sarvede rakkude ja ajutüve motoorsete tuumade atroofia (joon. 21-2), millele järgneb motoorsete närvide ja lihaskoe juurte atroofia. 
Riis. 21-3. Werdnig-Hoffmanni tõvega vastsündinu tüüpiline kehahoiak.
Haigus algab enne 2-aastaseks saamist, kuid kõige sagedamini sünnieelsel perioodil. On teateid üsna harva esinevatest sarnastest haigustest vanematel lastel. Selle varased ilmingud hõlmavad käte ja jalgade proksimaalsete ja distaalsete osade lihaste nõrkust ja hüpotensiooni, interkostaalset, innerveeritud kraniaalnärve. Lapse jalad on tüüpilises konnaasendis: puusast eraldatud ja põlveliigestest painutatud (joon. 21-3). Diafragma kannatab suhteliselt harva. Roietevaheliste lihaste nõrkusest tingitud hingamishäired väljenduvad selle paradoksaalsuses koos rindkere tagasitõmbumisega inspiratsiooni ajal. Välised silmalihased ei osale protsessis. Tavaliselt on märgatavad keelelihaste fibrillaarsed tõmblused. Kõõluste refleksid peaaegu alati puuduvad. Laste vaimne areng jääb normaalsesse vahemikku ning sisukas näoilme ja patsiendi tavapärane välimus on teravas vastuolus motoorse aktiivsuse puudumisega. Haiguse algstaadiumis on kalduvus ülekaalulisusele, hilisemates staadiumides ei saa patsiendid neelamisliigutusi teha. Surm võib tekkida hingamise seiskumise ja toidu aspireerimise tagajärjel. Kui haigus algab emakas, surevad lapsed tavaliselt enne 2-aastaseks saamist.
Hilisema algusega on oodatav eluiga mitu aastat, mõnikord elab patsient täiskasvanuks.
Werdnig-Hoffmanni tõve diagnoos põhineb suuresti kliinilistel tunnustel. Elektromüograafia andmed (fibrillatsioonid ja fastsikulaarsed tõmblused) näitavad lihaste denervatsiooni. Lihaskoe biopsia avastab degeneratsiooni eri staadiumides rakurühmad: iga lihaskiudude rühm sisaldab rakke, mida innerveerib üks motoorne neuron. CSF-i, närvijuhtivuse ja vereseerumis sisalduvate ensüümide aktiivsuse uurimisel patoloogiat ei tuvastata.
Eristage haigus paljudest vähem tüüpilistest seisunditest, mille puhul imikul on nõrkus ja hüpotensioon. Sel juhul nimetatakse seda loiuks (tabel 21-1).
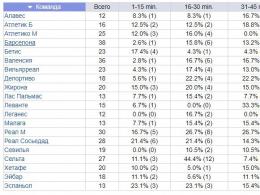
Tabel 21-1. Haigused, millega kaasneb püsiv lihaste hüpotensioon
Haigused
KNS | selgroog | perifeerne | närviliselt me | |
Atooniline | Polüneuriit (Guillain-Barré sündroom) | myasthenia gravis | kaasasündinud |
|
pärilik | Werdnig-Hoffmanni haigus | Perekond | Laste botulism | Müotooniline düstroofia |
Tuuma kollatõbi | Pärilik sensoorne neuropaatia | Glükogeeni ladestumise haigused vöötlihastes ja südamelihastes (Pompe tüüp) |
||
Kromosomaalne | Keskvarraste haigus |
|||
Okulotsebrorenaalne sündroom (Jloy) Aju lipidoos | Mitte karmiinpunane |
|||
Prader-Willi sündroom | päris |
Kesknärvisüsteemi düsfunktsiooni, millega kaasneb lihaste hüpotensioon, saab eristada perifeersest neuromuskulaarsest haigusest selliste tunnuste alusel nagu vähenenud reaktsioon visuaalsetele stiimulitele ja kõõluste reflekside säilimine. Werdnig-Hoffmanni tõbe perifeersete närvide ja lihaste haigustest saab mõnel juhul eristada alles pärast spetsiaalseid diagnostilisi meetodeid, nagu CSF-uuring, perifeersete närvide impulsi kiiruse ja seerumi ensüümide aktiivsuse määramine, lihaskoe biopsia. Siiski tuleb meeles pidada, et mõned hüpotooniliste häirete ilmingud lastel ei kuulu tabelis loetletud haiguste hulka. 21-1. Sellistes tingimustes säilib lihaste erutuvus, kõõluste refleksid on alla surutud, kuid tavaliselt ei kao need täielikult. Laboratoorsed uuringud, sealhulgas lihaskoe biopsia, ei tuvasta patoloogiat. Enamikul nende sümptomitega lastel kaovad hüpotensioon ja nõrkus järk-järgult. Nende iseloomustamiseks kasutatakse tavaliselt selliseid termineid nagu "healoomuline kaasasündinud hüpotensioon" ja "kaasasündinud amüotoonia". Siiski on kaheldav, et sellised sümptomid on homogeense haiguste rühma tunnused.
Juht
"Onkogeneetika"
Zhusina
Julia Gennadievna

Lõpetanud Voroneži Riikliku Meditsiiniülikooli pediaatriateaduskonna. N.N. Burdenko 2014. aastal.
2015 - teraapiapraktika Voroneži Riikliku Meditsiiniülikooli teaduskonnateraapia osakonna baasil. N.N. Burdenko.
2015 - sertifitseerimiskursus erialal "Hematoloogia" Moskva hematoloogiauuringute keskuse baasil.
2015-2016 – VGKBSMP nr 1 terapeut.
2016 - kinnitati arstiteaduste kandidaadi lõputöö teema "Haiguse kliinilise kulgemise ja prognoosi uurimine aneemilise sündroomiga kroonilise obstruktiivse kopsuhaigusega patsientidel". Rohkem kui 10 publikatsiooni kaasautor. Geneetika ja onkoloogia teaduslike ja praktiliste konverentside osaleja.
2017 - täiendkoolitus teemal: "pärilike haigustega patsientide geeniuuringute tulemuste tõlgendamine".
Alates 2017. aastast residentuuri erialal "Geneetika" RMANPE baasil.
Juht
"Geneetika"
Kanivets
Ilja Vjatšeslavovitš

Kanivets Ilja Vjatšeslavovitš, geneetik, meditsiiniteaduste kandidaat, meditsiinilise geenikeskuse Genomed geneetikaosakonna juhataja. Venemaa meditsiinilise täiendõppe akadeemia meditsiinigeneetika osakonna assistent.
Ta lõpetas 2009. aastal Moskva Riikliku Meditsiini- ja Stomatoloogiaülikooli arstiteaduskonna ning 2011. aastal residentuuri sama ülikooli meditsiinigeneetika osakonnas erialal "Geneetika". 2017. aastal kaitses ta arstiteaduste kandidaadi väitekirja teemal: DNA segmentide (CNV) koopiaarvu variatsioonide molekulaarne diagnostika kaasasündinud väärarengute, fenotüübi anomaaliate ja/või vaimse alaarenguga lastel SNP suure tihedusega oligonukleotiidide mikrokiipide abil. »
Aastatel 2011-2017 töötas ta Kliinilises Lastehaiglas geneetikuna. N.F. Filatov, föderaalse riigieelarvelise teadusasutuse "Meditsiinigeeniuuringute keskus" teaduslik nõustamisosakond. Aastast 2014 kuni praeguseni on ta juhtinud MHC Genomedi geneetikaosakonda.
Põhitegevused: pärilike haiguste ja kaasasündinud väärarengute, epilepsiaga patsientide diagnostika ja ravi, päriliku patoloogia või väärarengutega lapse sündinud perede meditsiinigeneetiline nõustamine, sünnieelne diagnostika. Konsultatsiooni käigus viiakse läbi kliiniliste andmete ja genealoogia analüüs, et selgitada välja kliiniline hüpotees ja vajalik kogus geenitesti. Küsitluse tulemuste põhjal tõlgendatakse andmeid ja selgitatakse saadud infot konsultantidele.
Ta on üks projekti Genetics School asutajatest. Esitab regulaarselt ettekandeid konverentsidel. Ta peab loenguid geneetikutele, neuroloogidele ja sünnitusarstidele-günekoloogidele, samuti pärilike haigustega patsientide vanematele. Ta on enam kui 20 artikli ja arvustuse autor ja kaasautor Venemaa ja välisajakirjades.
Erialaste huvide valdkond on kaasaegsete genoomi hõlmavate uuringute juurutamine kliinilisse praktikasse, nende tulemuste tõlgendamine.
Vastuvõtu aeg: K, R 16-19
Juht
"Neuroloogia"
Sharkov
Artem Aleksejevitš
Šarkov Artjom Aleksejevitš- neuroloog, epileptoloog
2012. aastal õppis ta Lõuna-Koreas Daegu Haanu ülikoolis rahvusvahelise programmi “Idamaine meditsiin” raames.
Alates 2012. aastast - osalemine xGenCloudi geneetiliste testide tõlgendamise andmebaasi ja algoritmi korraldamises (http://www.xgencloud.com/, projektijuht - Igor Ugarov)
2013. aastal lõpetas ta N. I. nimelise Venemaa riikliku teadusuuringute meditsiiniülikooli pediaatriateaduskonna. Pirogov.
Aastatel 2013–2015 õppis ta föderaalses riigieelarvelises teadusasutuses "Neuroloogia teaduskeskus" neuroloogia kliinilises residentuuris.
Alates 2015. aastast töötab ta neuroloogina, teadurina akadeemik Yu.E. nimelises Pediaatria Teadusliku Uurimise Kliinilises Instituudis. Veltishchev GBOU VPO RNIMU neid. N.I. Pirogov. Samuti töötab ta neuroloogina ja arstina A.I nimelise Epileptoloogia- ja Neuroloogiakeskuse kliinikute video-EEG monitooringu laboris. A.A. Ghazaryan“ ja „Epilepsiakeskus“.
2015. aastal õppis ta Itaalias koolis "2nd International Residential Course on Drug Resistant Epilepsies, ILAE, 2015".
2015. aastal täiendkoolitus - "Kliiniline ja molekulaargeneetika praktiseerivatele arstidele", RCCH, RUSNANO.
2016. aastal täienduskoolitus - "Molekulaargeneetika alused" bioinformaatika juhendamisel, Ph.D. Konovalova F.A.
Alates 2016. aastast - labori "Genomed" neuroloogilise suuna juhataja.
2016. aastal õppis ta Itaalias koolis "San Servolo rahvusvaheline edasijõudnute kursus: Brain Exploration and Epilepsy Surger, ILAE, 2016".
2016. aastal täienduskoolitus - "Innovaatilised geenitehnoloogiad arstidele", "Laborimeditsiini instituut".
Aastal 2017 - kool "NGS meditsiinigeneetikas 2017", Moskva Riiklik Teaduskeskus
Praegu viib ta läbi teaduslikke uuringuid epilepsia geneetika valdkonnas professori MD juhendamisel. Belousova E.D. ja professor, d.m.s. Dadali E.L.
Kinnitati meditsiiniteaduste kandidaadi lõputöö teema "Varajaste epilepsia entsefalopaatia monogeensete variantide kliinilised ja geneetilised omadused".
Peamisteks tegevusvaldkondadeks on epilepsia diagnoosimine ja ravi lastel ja täiskasvanutel. Kitsas spetsialiseerumine - epilepsia kirurgiline ravi, epilepsia geneetika. Neurogeneetika.
Teaduslikud publikatsioonid
Sharkov A., Sharkova I., Golovteev A., Ugarov I. "Diferentsiaaldiagnostika optimeerimine ja geneetilise testimise tulemuste tõlgendamine XGenCloudi ekspertsüsteemiga teatud epilepsiavormide korral". Meditsiiniline geneetika, nr 4, 2015, lk. 41.
*
Sharkov A.A., Vorobjov A.N., Troitski A.A., Savkina I.S., Dorofejeva M.Yu., Melikyan A.G., Golovteev A.L. "Epilepsia operatsioon multifokaalsete ajukahjustuste korral tuberoosskleroosiga lastel." XIV Venemaa kongressi "INNOVATIIVSED TEHNOLOOGIAD PEDIAATRIAS JA LASTEKIRURGIAS" kokkuvõtted. Vene perinatoloogia ja pediaatria bülletään, 4, 2015. - lk 226-227.
*
Dadali E.L., Belousova E.D., Sharkov A.A. "Molekulaargeneetilised lähenemisviisid monogeense idiopaatilise ja sümptomaatilise epilepsia diagnoosimiseks". XIV Venemaa kongressi "INNOVATIIVSED TEHNOLOOGIAD LASTE- JA LASTEKIIRURGIAS" kokkuvõte. Vene perinatoloogia ja pediaatria bülletään, 4, 2015. - lk 221.
*
Sharkov A.A., Dadali E.L., Sharkova I.V. "II tüüpi varajase epileptilise entsefalopaatia haruldane variant, mis on põhjustatud CDKL5 geeni mutatsioonidest meespatsiendil." Konverents "Epileptoloogia neuroteaduste süsteemis". Konverentsi materjalide kogumik: / Toimetanud: prof. Neznanova N.G., prof. Mihhailova V.A. Peterburi: 2015. - lk. 210-212.
*
Dadali E.L., Sharkov A.A., Kanivets I.V., Gundorova P., Fominykh V.V., Sharkova I.V. Troitski A.A., Golovtejev A.L., Poljakov A.V. KCTD7 geeni mutatsioonidest põhjustatud 3. tüüpi müokloonuse epilepsia uus alleelvariant // Meditsiiniline geneetika.-2015.- v.14.-№9.- lk.44-47
*
Dadali E.L., Sharkova I.V., Sharkov A.A., Akimova I.A. "Päriliku epilepsia diagnoosimise kliinilised ja geneetilised tunnused ja kaasaegsed meetodid". Materjalide kogumik "Molekulaarbioloogilised tehnoloogiad meditsiinipraktikas" / Toim. vastav liige RANEN A.B. Maslennikova.- Teema. 24.- Novosibirsk: Academizdat, 2016.- 262: lk. 52-63
*
Belousova E.D., Dorofeeva M.Yu., Sharkov A.A. Epilepsia tuberoosskleroosi korral. Raamatus "Ajuhaigused, meditsiinilised ja sotsiaalsed aspektid", toimetanud Gusev E.I., Gekht A.B., Moskva; 2016; lk.391-399
*
Dadali E.L., Sharkov A.A., Sharkova I.V., Kanivets I.V., Konovalov F.A., Akimova I.A. Pärilikud haigused ja sündroomid, millega kaasnevad palavikukrambid: kliinilised ja geneetilised omadused ning diagnostikameetodid. //Vene laste neuroloogia ajakiri.- T. 11.- nr 2, lk. 33-41. doi: 10.17650/ 2073-8803-2016-11-2-33-41
*
Sharkov A.A., Konovalov F.A., Sharkova I.V., Belousova E.D., Dadali E.L. Molekulaargeneetilised lähenemisviisid epilepsia entsefalopaatia diagnoosimiseks. Kokkuvõtete kogumik "VI BALTIC CONGRESS ON LASTE NEUROLOOGIA" / Toimetanud professor Guzeva V.I. Peterburi, 2016, lk. 391
*
Hemisferotoomia ravimiresistentse epilepsia korral kahepoolse ajukahjustusega lastel Zubkova N.S., Altunina G.E., Zemlyansky M.Yu., Troitsky A.A., Sharkov A.A., Golovteev A.L. Kokkuvõtete kogumik "VI BALTIC CONGRESS ON LASTE NEUROLOOGIA" / Toimetanud professor Guzeva V.I. Peterburi, 2016, lk. 157.
*
*
Artikkel: Varajase epilepsia entsefalopaatia geneetika ja diferentseeritud ravi. A.A. Sharkov*, I.V. Sharkova, E.D. Belousova, E.L. Dadali. Journal of Neurology and Psychiatry, 9, 2016; Probleem. 2doi:10.17116/jnevro20161169267-73
*
Golovteev A.L., Sharkov A.A., Troitski A.A., Altunina G.E., Zemljanski M.Yu., Kopatšev D.N., Dorofejeva M.Yu. "Epilepsia kirurgiline ravi tuberoosskleroosis", toimetanud Dorofeeva M.Yu., Moskva; 2017; lk.274
*
Epilepsiavastase rahvusvahelise liiga uued epilepsia ja epilepsiahoogude rahvusvahelised klassifikatsioonid. Journal of Neurology and Psychiatry. C.C. Korsakov. 2017. V. 117. nr 7. S. 99-106
Osakonna juhataja
"Eelsoodumuste geneetika",
bioloog, geneetiline konsultant
Dudurich
Vasilisa Valerievna

- "Eelsoodumuste geneetika" osakonna juhataja, bioloog, geneetiline konsultant
Aastal 2010 - PR-spetsialist, Kaug-Ida rahvusvaheliste suhete instituut
Aastal 2011 - Kaug-Ida föderaalülikooli bioloog
Aastal 2012 - Venemaa FGBUN SRI FCM FMBF "Genodiagnostika kaasaegses meditsiinis"
Aastal 2012 - Uuring "Geneetilise testimise juurutamine üldkliinikus"
Aastal 2012 - Erialane koolitus "Sünnieelne diagnostika ja geneetiline pass - ennetava meditsiini alus nanotehnoloogia ajastul" D.I.
Aastal 2013 - Bakulevi südame-veresoonkonna kirurgia teaduskeskuse erialakoolitus "Geneetika kliinilises hemostasioloogias ja hemorheoloogias"
Aastal 2015 - Erialane koolitus Venemaa Meditsiinigeneetika Seltsi VII kongressi raames
Aastal 2016 - Andmeanalüüsi kool "NGS meditsiinipraktikas" FGBNU "MGNTS"
Aastal 2016 - praktika "Geneetiline nõustamine" FGBNU "MGNTS"
Aastal 2016 - osales rahvusvahelisel inimgeneetika kongressil Kyotos, Jaapanis
Aastatel 2013-2016 - Habarovski meditsiinilise geneetikakeskuse juhataja
Aastatel 2015-2016 - Kaug-Ida Riikliku Meditsiiniülikooli bioloogia osakonna lektor
Aastatel 2016-2018 - Venemaa Meditsiinigeneetika Seltsi Habarovski filiaali sekretär
2018. aastal – võtsin osa seminarist "Venemaa reproduktiivpotentsiaal: versioonid ja vastuversioonid" Sotšis, Venemaa
Kool-seminari "Geneetika ja bioinformaatika ajastu: interdistsiplinaarne lähenemine teaduses ja praktikas" korraldaja - 2013, 2014, 2015, 2016
Geneetikakonsultandi kogemus - 7 aastat
Tsaritsa Alexandra heategevusfondi asutaja, et aidata geneetilise patoloogiaga lapsi alixfond.ru
Erialaste huvide valdkond: mürobioom, multifaktoriaalne patoloogia, farmakogeneetika, nutrigeneetika, reproduktiivgeneetika, epigeneetika.
Juht
"Sünnieelne diagnoos"
Kiiev
Julia Kirillovna

2011. aastal lõpetas ta Moskva Riikliku Meditsiini- ja Stomatoloogiaülikooli. A.I. Evdokimova üldmeditsiini erialal Õppinud residentuuris sama ülikooli meditsiinigeneetika osakonnas geneetika erialal
2015. aastal läbis ta praktika sünnitusabi ja günekoloogia erialal föderaalse riigieelarvelise kutsekõrgkooli MGUPP meditsiinilise kraadiõppe meditsiiniinstituudis.
Alates 2013. aastast on ta läbi viinud konsultatiivseid kohtumisi pereplaneerimise ja paljunemise keskuses DZM.
Alates 2017. aastast on ta Genomedi labori sünnieelse diagnostika osakonna juhataja
Esitab regulaarselt ettekandeid konverentsidel ja seminaridel. Loeb loenguid erinevate erialade arstidele reproduktiiv- ja sünnieelse diagnostika valdkonnas
Viib läbi meditsiinilist geeninõustamist rasedatele sünnieelse diagnostika alal, et vältida kaasasündinud väärarengutega laste sündi, samuti eeldatavalt pärilike või kaasasündinud patoloogiatega peredele. Viib läbi DNA diagnostika saadud tulemuste tõlgendamise.
SPETSIALISTID
Latypov
Artur Šamilevitš

Latypov Artur Šamilevitš – kõrgeima kvalifikatsioonikategooria arst geneetik.
Pärast Kaasani Riikliku Meditsiiniinstituudi arstiteaduskonna lõpetamist 1976. aastal töötas ta aastaid esmalt arstina meditsiinigeneetika kabinetis, seejärel Tatarstani Vabariikliku Haigla meditsiinigeneetikakeskuse juhatajana, meditsiinigeneetika peaspetsialistina. Tatarstani Vabariigi tervishoiuministeerium, Kaasani Meditsiiniülikooli osakondade õpetaja.
Enam kui 20 reproduktiiv- ja biokeemilise geneetika probleeme käsitleva teadusliku artikli autor, osalenud paljudel kodumaistel ja rahvusvahelistel meditsiinigeneetika probleeme käsitlevatel kongressidel ja konverentsidel. Ta tutvustas keskuse praktilisse töösse rasedate ja vastsündinute massilise pärilike haiguste sõeluuringu meetodeid, viis läbi tuhandeid invasiivseid protseduure loote pärilike haiguste kahtluse korral erinevates raseduse staadiumides.
Alates 2012. aastast töötab ta meditsiinigeneetika osakonnas sünnieelse diagnostika kursusega Venemaa kraadiõppeakadeemias.
Teadusvaldkonnad – laste ainevahetushaigused, sünnieelne diagnostika.
Vastuvõtuaeg: K 12-15, L 10-14Arstid võetakse vastu kokkuleppel.
Geneetik
Gabelko
Deniss Igorevitš

2009. aastal lõpetas ta KSMU nimelise arstiteaduskonna. S. V. Kurashova (eriala "Meditsiin").
Praktika Peterburi Föderaalse Tervise ja Sotsiaalarengu Agentuuri kraadiõppe meditsiiniakadeemias (eriala "Geneetika").
Praktika teraapias. Esmane ümberõpe erialal "Ultraheli diagnostika". Alates 2016. aastast on ta Fundamentaalmeditsiini ja Bioloogia Instituudi kliinilise meditsiini aluste osakonna osakonna töötaja.
Erialased huvid: sünnieelne diagnostika, kaasaegsete sõeluuringute ja diagnostikameetodite kasutamine loote geneetilise patoloogia tuvastamiseks. Pärilike haiguste kordumise riski määramine perekonnas.
Geneetika ning sünnitusabi ja günekoloogia teaduslike ja praktiliste konverentside osaleja.
Töökogemus 5 aastat.
Konsultatsioon kokkuleppelArstid võetakse vastu kokkuleppel.
Geneetik
Grishina
Christina Aleksandrovna

2015. aastal lõpetas ta Moskva Riikliku Meditsiini- ja Stomatoloogiaülikooli üldmeditsiini erialal. Samal aastal astus ta residentuuri föderaalsesse riigieelarvelisesse teadusasutusse "Meditsiinigeeniuuringute keskus" erialale 30.08.30 "Geneetika".
Ta võeti 2015. aasta märtsis tööle kompleksselt pärilike haiguste molekulaargeneetika laboratooriumisse (juhataja – bioloogiateaduste doktor Karpukhin A.V.) labori assistendiks. Alates 2015. aasta septembrist on ta üle viidud teaduri ametikohale. Ta on enam kui 10 kliinilist geneetikat, onkogeneetikat ja molekulaaronkoloogiat käsitleva artikli ja kokkuvõtte autor ja kaasautor Venemaa ja välismaistes ajakirjades. Regulaarne meditsiinigeneetika konverentside osaleja.
Teaduslike ja praktiliste huvide valdkond: päriliku sündroomi ja multifaktoriaalse patoloogiaga patsientide meditsiiniline geneetiline nõustamine.
Konsultatsioon geneetikuga võimaldab teil vastata järgmistele küsimustele:
Kas lapse sümptomid on päriliku haiguse tunnused?
milliseid uuringuid on vaja põhjuse tuvastamiseks
täpse prognoosi määramine
soovitused sünnieelse diagnoosi läbiviimiseks ja tulemuste hindamiseks
kõik, mida pead teadma pereplaneerimisest
IVF planeerimise konsultatsioon
väli- ja veebikonsultatsioonid
Geneetik
Gorgisheli
Ketevan Vazhaevna

Ta on lõpetanud N. I. nimelise Venemaa riikliku teadusuuringute meditsiiniülikooli meditsiini- ja bioloogiateaduskonna. Pirogov kaitses 2015. aastal väitekirja teemal "Keha seisundi elutähtsate näitajate kliiniline ja morfoloogiline korrelatsioon ning vere mononukleaarsete rakkude morfoloogilised ja funktsionaalsed omadused raske mürgistuse korral". Ta on lõpetanud eelnimetatud ülikooli molekulaar- ja rakugeneetika osakonna kliinilise residentuuri erialal "Geneetika".
võttis osa teaduslik-praktilisest koolist "Innovatiivsed geenitehnoloogiad arstidele: rakendamine kliinilises praktikas", Euroopa Inimgeneetika Ühingu (ESHG) konverentsist ja teistest inimgeneetikale pühendatud konverentsidest.
Viib läbi eeldatavalt pärilike või kaasasündinud patoloogiatega, sh monogeensete haiguste ja kromosoomianomaaliatega peredele meditsiinigeneetikanõustamist, määrab laboratoorsete geeniuuringute näidustused, tõlgendab DNA diagnostika tulemusi. Nõustab rasedaid sünnieelse diagnostika osas, et vältida kaasasündinud väärarengutega laste sündi.
Geneetik, sünnitusarst-günekoloog, meditsiiniteaduste kandidaat
Kudrjavtseva
Jelena Vladimirovna

Geneetik, sünnitusarst-günekoloog, meditsiiniteaduste kandidaat.
Reproduktiivnõustamise ja päriliku patoloogia valdkonna spetsialist.
Lõpetas 2005. aastal Uurali Riikliku Meditsiiniakadeemia.
Sünnitusabi ja günekoloogia residentuur
Praktika erialal "Geneetika"
Professionaalne ümberõpe erialal "Ultraheli diagnostika"
Tegevused:
- Viljatus ja raseduse katkemine Vasilisa Jurjevna

Ta on lõpetanud Nižni Novgorodi Riikliku Meditsiiniakadeemia arstiteaduskonna (eriala "Meditsiin"). Ta lõpetas FBGNU "MGNTS" kliinilise internatuuri kraadiga "geneetika". 2014. aastal läbis ta praktika emaduse ja lapsepõlve kliinikus (IRCCS materno infantile Burlo Garofolo, Trieste, Itaalia).
Alates 2016. aastast töötab ta Genomed LLC-s arsti konsultandina.
Osaleb regulaarselt geneetikaalastel teaduslikel ja praktilistel konverentsidel.
Põhitegevused: Konsulteerimine geneetiliste haiguste kliinilise ja laboratoorse diagnostika ning tulemuste tõlgendamise alal. Päriliku patoloogia kahtlusega patsientide ja nende perekondade ravi. Konsulteerimine raseduse planeerimisel, samuti raseduse ajal sünnieelse diagnostika osas, et vältida kaasasündinud patoloogiaga laste sündi.
Aastatel 2013–2014 töötas ta Rostovi Vähiuuringute Instituudi molekulaaronkoloogia laboris nooremteadurina.
Aastal 2013 - täiendkoolitus "Kliinilise geneetika aktuaalsed küsimused", Venemaa Tervishoiuministeeriumi Riiklik Meditsiiniülikool Rost Riiklik Kutsekõrgharidusasutus.
Aastal 2014 - täiendkoolitus "Reaalajas PCR-meetodi rakendamine somaatiliste mutatsioonide geenidiagnostikas", FBSI "Rospotrebnadzori epidemioloogia keskinstituut".
Alates 2014. aastast Rostovi Riikliku Meditsiiniülikooli meditsiinigeneetika laboratooriumi geneetik.
2015. aastal kinnitas ta edukalt "meditsiinilabori teaduri" kvalifikatsiooni. Ta on Austraalia meditsiiniteadlaste instituudi aktiivne liige.
2017. aastal - täiendkoolitus "Pärilike haigustega patsientide geeniuuringute tulemuste tõlgendamine", NOCHUDPO "Arsti- ja farmaatsia täiendõppe koolituskeskus"; "Kliinilise laboridiagnostika ja laboratoorse geneetika tegelikud küsimused", Venemaa Tervishoiuministeeriumi Rostovi Riikliku Meditsiiniülikooli föderaalne eelarveline kõrgharidusasutus; täiendkoolitus "BRCA Liverpool Genetic Counseling Course", Liverpooli Ülikool.
Osaleb regulaarselt teaduskonverentsidel, on enam kui 20 teaduspublikatsiooni autor ja kaasautor kodu- ja välisväljaannetes.
Põhitegevus: DNA diagnostika tulemuste kliiniline ja laboratoorne tõlgendamine, kromosoomide mikrokiibi analüüs, NGS.
Huviala: uusimate kogu genoomi hõlmavate diagnostikameetodite rakendamine kliinilises praktikas, onkogeneetika.
Pärilikud neuromuskulaarsed haigused on suur heterogeenne rühm haigusi, mis põhinevad neuromuskulaarse aparaadi geneetiliselt määratud kahjustusel. Haigustele on iseloomulik lihasnõrkus, lihaste atroofia, staatilise ja liikumisfunktsiooni häired.
Diagnoosimisel võetakse haiguse esimeste kliiniliste sümptomite ilmnemise vanus, atroofia lokaliseerimine ja müodüstroofilise protsessi leviku olemus (tõusev, laskuv, pseudohüpertroofia olemasolu või puudumine, fascikulatsioonid, tundlikkushäired, paroksüsmid). lihasnõrkust), samuti voolukiirust.
Progresseeruvad lihasdüstroofiad on kõige ulatuslikum rühm. Sõltuvalt primaarsete muutuste olemusest eristatakse tinglikult progresseeruvate lihasdüstroofiate primaarsed (müopaatiad) ja sekundaarsed vormid (denervatsiooni amüotroofiad - seljaaju ja neuraalsed).
24.1.1. progresseeruvad lihasdüstroofiad
Müodüstroofiate põhjuste selgitamiseks on välja pakutud mitmeid hüpoteese (neurogeensed, vaskulaarsed, membraanid), mis käsitlevad progresseeruvate lihasdüstroofiate mehhanisme primaarse, geneetiliselt määratud defekti seisukohast.
Progresseeruv Duchenne'i lihasdüstroofia. Seda haigust kirjeldas Duchenne 1853. aastal. Esinemissagedus on 3,3 juhtu 100 000 elaniku kohta, 14 juhtu 100 000 sündinu kohta. See on päritud retsessiivse, X-seotud tüübina. Enamikul juhtudel on poisid haiged. Duchenne'i düstroofia on seotud düstrofiini tootmise eest vastutava geeni kahjustusega. Uurides emasid - geenikandjaid geenikonsultatsioonidel (koorioni villi biopsia 8.-9. nädalal), avastatakse haigus poistel. Tüdrukute haigusjuhud on äärmiselt haruldased, kuigi need on võimalikud karüotüübi X0, X0 / XX, X0 / XXX, X0 / XXX / XXX mosaiiksuse ja kromosoomide struktuursete kõrvalekallete korral.
Patomorfoloogia. Seda iseloomustab lihaskoe degeneratsioon, selle asendamine rasv- ja sidekoega ning üksikute kiudude nekroos.
Kliinilised ilmingud. Haiguse sümptomid ilmnevad esimesel 1-3 eluaastal. Juba 1. kursusel köidab tähelepanu laste mahajäämus motoorses arengus. Nad hakkavad reeglina maha istuma, tõusma, kõndima viivitusega. Liigutused on kohmakad, kõndides on lapsed ebastabiilsed, sageli komistavad, kukuvad. 2-3-aastaselt ilmneb lihasnõrkus, patoloogiline lihasväsimus, mis väljendub füüsilisel pingutusel – pikal kõndimisel, trepist ronimisel, kõnnaku muutumisel nagu "pardil". Sel perioodil juhitakse tähelepanu laste liigutuste omapärasele "stereotüüpsele" dünaamikale horisontaalasendist, kükiasendist või toolilt püsti tõusmisel. Püsti tõusmine toimub etapiviisiliselt, aktiivse käte kasutamisega – „redelil ronimine“ või „ise ronimine“. Lihaste atroofia on alati sümmeetriline. Esialgu lokaliseeritakse need alajäsemete proksimaalsetes lihasrühmades - vaagnavöötme lihastes, reites ja 1-3 aasta pärast levivad nad kiiresti ülespoole ülemiste jäsemete proksimaalsetesse lihasrühmadesse - õlavöötmesse. , seljalihased. Atroofia tagajärjel tekivad lordoos, "pterügoidne" abaluud ja "herilase" vöökoht. Tüüpiline, "klassikaline" haiguse sümptom on säärelihaste pseudohüpertroofia.
Palpatsioonil on lihased tihedad, valutud. Paljudel patsientidel tekivad erinevate lihasrühmade selektiivsete ja ebaühtlaste kahjustuste tagajärjel varakult lihaskontraktuurid ja kõõluste tagasitõmbed. Lihastoonus väheneb peamiselt proksimaalsetes lihasrühmades. Kõõluste refleksid muutuvad erineva järjestusega. Haiguse algfaasis kaovad põlverefleksid, hiljem - biitsepsi- ja triitsepsilihaste refleksid. Kalcaneaalsed (Achilleuse) refleksid jäävad pikka aega puutumatuks.
Vähenenud võnkeamplituud ja suurenenud mitmefaasilisus.
Üks Duchenne'i müodüstroofia eristavaid tunnuseid on selle vormi kombinatsioon osteoartikulaarse süsteemi ja siseorganite (kardiovaskulaarsed ja neuroendokriinsed süsteemid) patoloogiaga. Osteoartikulaarseid häireid iseloomustavad lülisamba, jalgade ja rinnaku deformatsioonid. Röntgenpiltidel leitakse luuüdi kanali ahenemine, toruluude pikkade diafüüside kortikaalse kihi hõrenemine.
Südame-veresoonkonna häired ilmnevad kliiniliselt pulsi labiilsusest, vererõhust, mõnikord ka toonuste kurtusest ja südame piiride laienemisest. EKG-l registreeritakse muutused müokardis (His-kimbu jalgade blokaad jne). Neuroendokriinsed häired esinevad 30-50% patsientidest. Teistest sagedamini täheldatakse Itsenko-Cushingi sündroomi, Babinsky-Frelichi adiposogenitaalset düstroofiat. Paljude patsientide intelligentsus väheneb erineval määral.
Voolu. Haigusel on kiiresti progresseeruv pahaloomuline kulg. 7-10. eluaastaks tekivad süvaliikumise häired – väljendunud kõnnaku muutus, lihasjõu vähenemine, mis piiravad suuresti patsientide vaba, iseseisvat liikumist. 14-15-aastaselt tekib liikumatus.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogiliste analüüside andmete (retsessiivne X-seotud pärilikkuse tüüp), haiguse kliiniliste ägenemiste (varajane algus 1-3 aastaselt, proksimaalsete lihasrühmade sümmeetriline atroofia, mis areneb ülespoole, pseudohüpertroofia) põhjal. vasika lihased, rasked somaatilised ja neuroendokriinsed häired, intelligentsuse langus, haiguse kiire pahaloomuline kulg), biokeemilised andmed (tavaliselt varakult, alates lapse 5. elupäevast, CPK aktiivsuse tõus - 30-50 korda kõrgem kui tavaliselt) , nõelelektromüograafia ja morfoloogilised tulemused. mis võimaldab tuvastada primaarset lihaste (müodüstroofset) tüüpi kahjustust.
Seda haigust tuleks eristada Werdnig-Hoffmanni seljaaju amüotroofiast, rahhiidist, puusa kaasasündinud nihestusest.
Beckeri progresseeruv lihasdüstroofia. Haigust kirjeldas Becker 1955. aastal. Esinemissagedus pole täpselt kindlaks tehtud. See on päritud retsessiivse X-seotud tüübina.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad 10-15-aastaselt, mõnikord ka varem. Esialgseteks sümptomiteks on lihasnõrkus, patoloogiline lihaste väsimus treeningu ajal, säärelihaste pseudohüpertroofia. Atroofiad arenevad sümmeetriliselt. Esialgu lokaliseeritakse need alajäsemete proksimaalsetes lihasrühmades - vaagnavöötmes ja reitel ning seejärel levivad ülemiste jäsemete proksimaalsetesse lihasrühmadesse. Atroofia tagajärjel tekivad muutused "pardi" tüüpi kõnnakus, püstitõusmisel kompenseerivad müopaatilised võtted. Proksimaalsete lihasrühmade lihastoonus on mõõdukalt vähenenud. Kõõluste refleksid püsivad pikka aega puutumata, ainult põlvetõmblused vähenevad varakult. Kardiovaskulaarsed häired on mõõdukalt väljendunud. Mõnikord on kardialgia, tema kimbu jalgade blokaad. Endokriinsüsteemi häired väljenduvad günekomastias, libiido languses, impotentsuses. Intelligentsus on salvestatud.
Voolu. Haigus areneb aeglaselt. Atroofia leviku kiirus on madal ja patsiendid püsivad pikka aega funktsionaalsed.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi põhjal (X-kromosoomiga seotud retsessiivne pärand), kliiniliste ilmingute tunnuste (haiguse algus 10-15-aastaselt, proksimaalsete lihasrühmade atroofia, aeglane, üle 10-aastased) põhjal. 20 aastat, atroofia levik ülespoole, massiivne gastrocnemius lihaste pseudohüpertroofia, mõõdukad somaatilised häired, aeglane kulg), biokeemilised andmed (CPK, LDH suurenenud aktiivsus veres), nõelelektromüograafia ja morfoloogilised tulemused, mis paljastavad esmase haiguse. lihaste tüüpi muutused.
Seda haigust tuleks eristada Duchenne'i, Erba-Rothi progresseeruvatest lihasdüstroofiatest ja Kugelberg-Welanderi spinaalsest amüotroofiast.
Progresseeruv Dreyfuse lihasdüstroofia. Haigust kirjeldas Dreyfus 1961. aastal. Esinemissagedus pole kindlaks tehtud. See on päritud retsessiivse X-seotud tüübina.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad 5-7-aastaselt. Nagu ka teiste progresseeruvate lihasdüstroofia vormide puhul, iseloomustab haiguse algust lihasnõrkus, patoloogiline lihaste väsimus treeningu ajal. Atroofiad tekivad sümmeetriliselt ja paiknevad algselt alajäsemete proksimaalsetes lihasrühmades - vaagnavöötmes, reitel. Ülemiste jäsemete proksimaalsed lihasrühmad osalevad müodüstroofilises protsessis palju hiljem. Selle vormi iseloomulikud tunnused on küünarliigeste varased kontraktuurid, Achilleuse kõõluste tagasitõmbumine. Paljudel patsientidel on südame rütmihäired. Intelligentsus on salvestatud.
Voolu. Haigus areneb aeglaselt.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi (retsessiivne X-seotud pärilikkuse tüüp), kliiniliste tunnuste (haiguse algus 5-7-aastaselt, sümmeetrilised atroofiad esialgse lokaliseerumisega proksimaalsetes alumistes lihasrühmades ja hiljem koos alumiste lihasrühmadega) põhjal. müodüstroofia aeglane levik ülemiste jäsemete proksimaalsetesse lihasrühmadesse, küünarliigeste varajased kontraktuurid, Achilleuse kõõluste tagasitõmbed, südame-veresoonkonna häired südame rütmihäirete kujul, aeglane, progresseeruv kulg), biokeemilised andmed (kõrge CPK aktiivsus) , elektromüograafia ja morfoloogilised andmed, mis näitavad esmaseid lihase iseloomu muutusi.
Haigust tuleks eristada Beckeri, Duchenne'i, Erba-Rothi, Kugelberg-Welanderi seljaaju amüotroofia progresseeruvatest lihasdüstroofiatest.
Erb-Rothi progresseeruv lihasdüstroofia. Sagedus 1,5 100 000 elaniku kohta. See on päritud autosoomselt retsessiivsel viisil.
Patomorfoloogia. Vastab esmasele lihaskahjustusele.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad peamiselt 14-16-aastaselt, üliharva - 5-10-aastaselt. Esialgseteks sümptomiteks on lihasnõrkus, patoloogiline lihaste väsimus füüsilisel pingutusel ja pardi tüüpi kõnnak. Atroofia haiguse alguses on lokaliseeritud alajäsemete proksimaalsetes lihasrühmades. Mõnikord mõjutab müodüstroofne protsess samaaegselt vaagna- ja õlavöötme lihaseid. Palju hilisemates etappides on protsessi kaasatud selja- ja kõhulihased. Atroofia tagajärjel ilmnevad lordoos, "pterygoid" abaluud, "herilane" vöökoht. Tõusmisel kasutavad patsiendid abivõtteid - "redeliga" tõusmist. Lihaste pseudohüpertroofia, liigeste kontraktuurid, kõõluste tagasitõmbed on reeglina mõõdukalt väljendunud. Juba haiguse algstaadiumis on tüüpiline põlvereflekside ja õla biitsepsi- ja triitsepsilihaste reflekside vähenemine.
Voolu. Haigus areneb kiiresti. Puue tuleb varakult.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi (autosoomne retsessiivne pärand), kliiniliste tunnuste (haiguse algus peamiselt 14-16-aastaselt, proksimaalsete lihasrühmade atroofia, mõõdukas pseudohüpertroofia, kiire progresseerumine), nõela tulemuste põhjal. elektromüograafia ja morfoloogilised andmed, mis võimaldavad tuvastada esmaseid lihasmuutusi.
Seda haigust tuleks eristada progresseeruvast Beckeri lihasdüstroofiast, Kugelberg-Welanderi seljaaju amüotroofiast.
Landouzy-Dejerine õla-näo vorm. Seda haigust kirjeldasid Landouzi ja Dejerine aastal 1884. Esinemissagedus on 0,9-2 juhtu 100 000 elaniku kohta. See on päritud autosomaalselt domineerival viisil.
Kliinilised ilmingud. Esimesed märgid ilmnevad peamiselt 10-20-aastaselt. Lihasnõrkus, atroofia lokaliseeritakse näo, abaluude, õlgade miimilistes lihastes. Atroofia tõttu muutub nägu hüpomiimiliseks. Tüüpilised on “poleeritud” otsmik, lagoftalmos, “põiki” naeratus, paksud, kohati keerdunud huuled (”tapir huuled”). Õla biitsepsi ja triitsepsi lihaste atroofia, rinnalihaste suur, eesmine serratus, trapetslihased põhjustavad vaba õlavöötme, "pterügoidsete" abaluude sümptomite ilmnemist, laia abaluudevahelise ruumi ilmnemist, rindkere lamenemist, skolioosi. Mõnel juhul laieneb atroofia ka jalalihastele (abaluu-õla-reieluu, näo-õla-õla-reieluu, näo-õla-tuhara-reieluu, näo-õla-õla-reieluu-peroneaal- ja muud variandid). Pseudohüpertroofia väljendub sääre- ja deltalihastes. Lihastoonus haiguse algfaasis väheneb proksimaalsetes lihasrühmades. Kõõluste refleksid vähenevad peamiselt õla biitsepsi ja triitsepsi lihastega.
Voolu. Reeglina areneb haigus aeglaselt. Patsiendid on pikka aega funktsionaalsed.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi (autosoomne dominantne pärand), kliiniliste tunnuste (peamiselt müodüstroofse protsessi humeroscapulaar-näo lokaliseerimine) põhjal.
Seda haigust tuleks eristada teistest progresseeruvatest lihasdüstroofiatest: Erba-Roth, Becker.
24.1.2. Neurogeensed amüotroofiad
Werdnig-Hoffmanni seljaaju amüotroofia. Seda haigust kirjeldasid J. Werdnig 1891. aastal ja J. Hoffmann 1893. aastal. Esinemissagedus on 1 juhtu 100 000 elaniku kohta, 7 juhtu 100 000 vastsündinu kohta. See on päritud autosoomselt retsessiivsel viisil.
Patomorfoloogia. Leitakse seljaaju eesmiste sarvede rakkude vähearengut, eesmiste juurte demüeliniseerumist. Sageli on sarnased muutused V, VI, VII, IX, X, XI ja XII kraniaalnärvide motoorsetes tuumades ja juurtes. Skeletilihastes iseloomustavad neurogeenseid muutusi “kimbu atroofia”, atroofeerunud ja tervete lihaskiudude kimpude vaheldumine, aga ka primaarsetele müopaatiatele omased häired (hüalinoos, üksikute lihaskiudude hüpertroofia, sidekoe hüperplaasia).
Kliinilised ilmingud. Haigusel on kolm vormi: kaasasündinud, varane lapsepõlv ja hiline, mis erinevad esimeste kliiniliste sümptomite ilmnemise aja ja amüotroofse protsessi kiiruse poolest.
Kaasasündinud vormis väljendub laste esimestest elupäevadest alates üldine lihaste hüpotoonia ja lihaste hüpotroofia, kõõluste reflekside vähenemine või puudumine. Bulbaarsed häired määratakse varakult, mis väljenduvad loid imemises, nõrgas nutmises, keele virvenduses ja neelu refleksi vähenemises. Haigus on kombineeritud osteoartikulaarsete deformatsioonidega: skolioos, lehtrikujuline või "kana" rind, liigeste kontraktuurid. Staatiliste ja lokomotoorsete funktsioonide areng on järsult aeglustunud. Ainult piiratud arvul lastel areneb suure hilinemisega võime oma pead kinni hoida ja iseseisvalt istuda. Omandatud motoorsed oskused aga taanduvad kiiresti. Paljudel lastel, kellel on kaasasündinud haigusvorm, on intelligentsus vähenenud. Sageli täheldatakse kaasasündinud väärarenguid: kaasasündinud vesipea, krüptorhidism, hemangioom, puusaliigese düsplaasia, lampjalgsus jne.
Voolu. Haigusel on kiiresti progresseeruv kulg. Surm saabub enne 9. eluaastat. Üks peamisi surmapõhjuseid on rasked somaatilised häired (südame-veresoonkonna ja hingamispuudulikkus), mis on põhjustatud rindkere lihaste nõrkusest ja selle osaluse vähenemisest hingamisfüsioloogias.
Varase lapsepõlvevormi korral ilmnevad esimesed haigusnähud reeglina elu teisel poolel. Esimeste kuude motoorne areng on rahuldav. Lapsed hakkavad õigeaegselt pead hoidma, istuma, mõnikord seisma. Haigus areneb alaägedalt, sageli pärast nakatumist, toidumürgitust. Lõtv parees lokaliseerub esialgu jalgades, seejärel levib kiiresti pagasiruumi ja käte lihastesse. Hajus lihaste atroofia on kombineeritud fastsikulatsioonide, keele virvenduse, sõrmede peene värisemise ja kõõluste kontraktuuridega. Lihastoonus, kõõluste ja perioste refleksid vähenevad. Hilisematel etappidel esineb üldistatud lihaste hüpotensioon, bulbarhalvatuse nähtused.
Voolu. Pahaloomuline, kuigi kergem kui kaasasündinud vorm. Surmav tulemus saabub 14-15-aastaselt.
Hilise vormi korral ilmnevad esimesed haigusnähud 1,5–2,5 aasta pärast. Selleks vanuseks on lastel staatiliste ja lokomotoorsete funktsioonide moodustumine täielikult lõppenud. Enamik lapsi kõnnib ja jookseb iseseisvalt. Haigus algab märkamatult. Liigutused muutuvad kohmakaks, ebakindlaks. Lapsed komistavad ja kukuvad sageli. Kõnnak muutub: nad kõnnivad kõverdatud põlvedega ("kellavärgi nuku" kõnnak). Lõtv parees lokaliseerub esialgu alajäsemete proksimaalsetes lihasrühmades, seejärel liigub suhteliselt aeglaselt ülemiste jäsemete proksimaalsetesse lihasrühmadesse, pagasiruumi lihastesse; lihaste atroofia on tavaliselt peen hästi arenenud nahaaluse rasvakihi tõttu. Tüüpilised fastsikulatsioonid, peentreemor sõrmedel, bulbar sümptomid - keele virvendus ja atroofia, neelu- ja palatine reflekside vähenemine. Kõõluste ja luuümbrise refleksid tuhmuvad juba haiguse varases staadiumis. Osteoartikulaarsed deformatsioonid arenevad paralleelselt põhihaigusega. Kõige väljendunud rindkere deformatsioon.
Voolu. Pahaloomuline, kuid pehmem kui kaks esimest vormi. Iseseisva kõndimise võime rikkumine toimub 10-12-aastaselt. Patsiendid elavad kuni 20-30 aastat.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos põhineb genealoogilisel analüüsil (autosoomne retsessiivne pärand), kliinilistel tunnustel (varajane algus, difuusne atroofia, mis paikneb peamiselt proksimaalsetes lihasrühmades, generaliseerunud lihaste hüpotensioon, keele fascikulatsioonid ja virvendused, pseudohüpertroofia puudumine, progredentne ja enamikul juhtudel pahaloomuline kulg jne), globaalse (naha) ja nõelelektromüograafia ning skeletilihaste morfoloogiliste uuringute tulemused, mis paljastavad muutuste denervatsiooni iseloomu.
Kaasasündinud ja varased vormid tuleks eristada eelkõige haigustest, mis kuuluvad kaasasündinud lihashüpotensiooniga sündroomide rühma (nn loid lapse sündroom): Oppenheimi amiatoonia, kaasasündinud healoomuline lihasdüstroofia vorm, tserebraalparalüüsi atooniline vorm, pärilikud ainevahetushaigused, kromosomaalsed sündroomid ja teised.Hilist vormi tuleks eristada Kugelberg-Welanderi spinaalsest amüotroofiast, Duchenne'i, Erb-Rothi progresseeruvatest lihasdüstroofiatest jne.
Ravi. Werdnig-Hoffmanni spinaalse amüotroofia korral on ette nähtud harjutusravi, massaaž, närvikoe trofismi parandavad ravimid - tserebrolüsiin, aminalon (gammalon), püriditool (entsefabool).
Spinaalne juveniilne Kugelberg-Welanderi pseudomüopaatiline lihasatroofia.
Patomorfoloogia. Leitakse seljaaju eesmiste sarvede rakkude alaareng ja degeneratsioon, eesmiste juurte demüelinisatsioon, IX, X, XII kraniaalnärvide motoorsete tuumade degeneratsioon. Skeletilihastes - kombineeritud muutused, mis on tüüpilised neurogeensele amüotroofiale (lihaskiudude fassikulaarne atroofia) ja primaarsele müodüstroofiale (lihaskiudude atroofia ja hüpertroofia, sidekoe hüperplaasia).
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad 4-8-aastaselt. Haiguse ilmnemise juhtumeid kirjeldatakse ka hilisemas vanuses - 15-30 aastat. Haiguse alguses on iseloomulikeks sümptomiteks jalgade patoloogiline lihaste väsimus pikaajalisel füüsilisel koormusel (kõndimine, jooksmine), mõnikord spontaansed lihastõmblused.
Väliselt köidavad tähelepanu laienenud säärelihased. Atroofiad paiknevad algselt alajäsemete proksimaalsetes lihasrühmades, vaagnavöötmes ja reites ning on alati sümmeetrilised. Nende välimus põhjustab jalgade motoorsete funktsioonide piiranguid - raskusi trepist ronimisel, horisontaalselt pinnalt tõusmisel. Kõnnak muutub järk-järgult. Selgete motoorsete häirete staadiumis omandab see "pardi" iseloomu. Ülemiste jäsemete proksimaalsete lihasrühmade atroofia areneb tavaliselt mitu aastat pärast alajäsemete lüüasaamist. Abaluu ja õlapiirkonna atroofia tõttu väheneb käte aktiivsete liigutuste maht, abaluud muutuvad "pterügoidseks". Proksimaalsete lihasrühmade lihastoonus väheneb. Kõõluste refleksid tuhmuvad kõigepealt jalgadel ja seejärel kätel (õla biitsepsi ja triitsepsi lihaste refleksid). Iseloomulikud sümptomid, mis eristavad Kugelberg-Welanderi spinaalset amüotroofiat Erb-Rothi fenotüüpiliselt sarnasest primaarsest progresseeruvast lihasdüstroofiast, on lihaste virvendus, keele virvendus ja sõrmede peen treemor. Luu-liigese häired, kõõluste tagasitõmbed on mõõdukalt väljendunud või puuduvad.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi andmete põhjal (autosoom-retsessiivne, autosoomdominantne, X-seotud retsessiivne pärilikkus), kliinilised tunnused (haigestumine on valdavalt 4-8-aastaselt, sümmeetriline lihasatroofia, levivad mööda tõusvat tüüpi lihasfaskulatsiooni, keele väike treemor, gastrocnemius lihaste pseudohüpertroofia, aeglane progresseeruv kulg), globaalse ja nõelelektromüograafia ning skeletilihaste morfoloogilise uuringu tulemused, mis paljastavad muutuste denervatsiooni iseloomu.
Seda haigust tuleks eristada Beckeri, Erba-Rothi, Werdnig-Hoffmanni seljaaju amüotroofia progresseeruvatest lihasdüstroofiatest.
Pärilik distaalne seljaaju amüotroofia. Sagedus pole määratud. See on päritud autosoomselt retsessiivselt, harvem autosoomselt dominantselt, X-seotud retsessiivselt.
Patomorfoloogia. Vastab teistele seljaaju amüotroofiale m.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad peamiselt esimesel elukümnendil. Haiguse esmased sümptomid on jalgade distaalsete lihaste nõrkus ja atroofia. 25% juhtudest täheldatakse käte distaalsete lihaste nõrkust ja atroofiat. Iseloomulikud tunnused - jalgade jämedad deformatsioonid, Achilleuse refleksi varajane kadumine põlveliigese ja käte sügavate reflekside säilimisega, sensoorsete häirete puudumine.
Voolu. Haigus areneb aeglaselt.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi (autosoomne dominantne, autosoomne retsessiivne, X-seotud retsessiivne pärilikkuse tüüp), kliiniliste tunnuste (algamine esimesel elukümnendil, atroofia domineeriv lokaliseerimine alajäsemete distaalsetes osades) põhjal, jalalaba jämedad deformatsioonid, sensoorsete häirete puudumine, müodüstroofse protsessi aeglane progresseerumine), globaalse ja nõelelektromüograafia tulemused, mis näitavad seljaaju eesmiste sarvede kaasamist protsessi.
Seda haigust tuleks eristada distaalsest Gowersi-Welanderi müopaatiast, Charcot-Marie-Toothi neuraalsest amüotroofiast.
Charcot-Marie-Toothi neuraalne amüotroofia. Esinemissagedus 1 50 000 elaniku kohta. See on päritud autosoomselt dominantselt, harvem autosoomselt retsessiivselt X-seotud mustriga.
Patomorfoloogia. Segmentaalne demüelinisatsioon leitakse närvides, lihastes - denervatsioon koos lihaskiudude "kiire" atroofia nähtustega.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad sageli 15-30-aastaselt, harvem koolieelses eas. Haiguse alguses on iseloomulikud sümptomid lihasnõrkus, patoloogiline väsimus alajäsemete distaalsetes osades. Patsiendid väsivad kiiresti ühes kohas pikaajalisest seismisest ja kasutavad sageli lihaste väsimuse vähendamiseks paigal kõndimist ("tallamise sümptom"). Harvemini algab haigus sensoorsete häiretega – valu, paresteesia, roomamine. Atroofiad arenevad esialgu säärte ja labajala lihastes. Lihaste atroofiad on tavaliselt sümmeetrilised. Mõjutatud on peroneaalne lihasrühm ja sääreluu eesmine lihas. Atroofia tõttu kitsenevad jalad distaalsetes osades järsult ja on "ümberpööratud pudelite" või "toonekurejalgade" kujul. Jalad on deformeerunud, muutuvad "sööduks", kõrge kaarega. Pareesitabel muudab patsientide kõnnakut. Kõnnivad jalad kõrgel: kandadel kõndimine on võimatu. Käte distaalsetes osades - siisar-, hüpotenaarlihastes, aga ka käte väikestes lihastes esinevad atroofiad ühinevad mitu aastat pärast amüotroofsete muutuste tekkimist jalgades. Käte atroofia on sümmeetriline. Rasketel juhtudel, raske atroofia korral, on käed "küüniste", "ahvide" kujul. Distaalsetes jäsemetes on lihastoonus ühtlaselt vähenenud. Kõõluste refleksid muutuvad ebaühtlaselt: Achilleuse refleksid vähenevad haiguse algstaadiumis ning põlverefleks, refleksid õla kolme- ja biitsepsi lihastest jäävad pikaks ajaks puutumata. Sensoorsed häired on määratletud perifeerset tüüpi pindmise tundlikkuse häiretega ("kinnaste ja sokkide tüüp"). Sageli esineb vegetatiivseid-troofilisi häireid - käte ja jalgade hüperhidroos ja hüperemia. Intelligentsus on tavaliselt säilinud.
Voolu. Haigus areneb aeglaselt. Prognoos on enamikul juhtudel soodne.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos põhineb genealoogiliste analüüside andmetel (autosoomne dominantne, autosoomne retsessiivne, X-seotud retsessiivne pärilikkus), kliinilised tunnused (distaalsete jäsemete atroofia, polüneuriitilist tüüpi tundlikkuse häired, aeglane progresseeruv kulg), globaalse, nõelaga ja stimulatsioonielektromüograafia (juhtimiskiiruse vähenemine mööda perifeersete närvide sensoorseid ja motoorseid kiude) ja mõnel juhul närvibiopsia.
Seda haigust tuleb eristada distaalsest Gowers-Welanderi müopaatiast, pärilikust distaalsest spinaalsest amüotroofiast, müotoonilisest düstroofiast, perifeersest neuropaatiast, mürgistusest, nakkuslikust polüneuriidist ja muudest haigustest.
Ravi. Progresseeruvate neuromuskulaarsete haiguste ravi eesmärk on parandada lihaste trofismi, samuti impulsside juhtivust piki närvikiude.
Trofismi parandamiseks määratakse hiirtele adenosiintrifosforhapet, kokarboksülaasi, tserebrolüsiini, riboksiini, fosfadeeni, karnitiinkloriidi, metnoniini, leutsiini, glutamiinhapet. Anaboolsed hormoonid on ette nähtud ainult lühikeste kursuste kujul. Kasutatakse vitamiine E, A, rühmad B ja C. Näidatud on mikrotsirkulatsiooni parandavaid vahendeid: nikotiinhape, ksantinoolnikotinaat, nikospan, pentoksifülliin, parmidiin. Juhtivuse parandamiseks on ette nähtud antikoliinesteraasi ravimid: galantamiin, oksasiil, püridostigmiinbromiid, stefaglabriinsulfaat, amüridiin.
Koos ravimteraapiaga kasutatakse harjutusravi. massaaž ja füsioteraapia. Oluline on jäsemete osteoartikulaarsete deformatsioonide ja kontraktuuride ennetamine.
Patsientide kompleksravis kasutatakse järgmisi füsioteraapia liike: ravimite elektroforees (prozeriin, kaltsiumkloriid), diadünaamilised voolud, müostimulatsioon sinusoidsete moduleeritud vooludega, närvide elektriline stimulatsioon, ultraheli, osokeriit, mudarakendused, radoon, okaspuu, sulfiid- ja vesiniksulfiidvannid, hapnikubaroteraapia. Ortopeediline ravi on näidustatud jäsemete kontraktuuride, mõõduka lülisamba deformatsiooni ja jäsemete asümmeetrilise lühenemise korral. Näidatakse täisväärtuslikke valke, kaaliumi dieeti, vitamiine.
Ravi peaks olema individuaalne, kompleksne ja pikaajaline, koosnema järjestikustest kursustest, sealhulgas erinevate teraapialiikide kombinatsioonist.
24.1.3. Paroksüsmaalne müopleegia
Pärilik paroksüsmaalne müopleegia on neuromuskulaarsete haiguste rühm, mida iseloomustavad äkilised lihasnõrkuse ja pleegiahood. Kõige levinumad pärilikud paroksüsmaalsed müoplegiad on hüpo-, hüper- ja normaleemilised vormid. Patogenees on ebaselge. Eeldatakse geneetiliselt määratud defekti sarkolemmaalses membraanis, mis halvendab naatriumi- ja kaaliumiioonide läbilaskvust,
Paroksüsmaalse müopleegia hüpokaleemiline vorm (Westfali tõbi). Haigust kirjeldas Westphal aastal 1895. See pärineb autosoomselt domineerivalt.
Kliinilised ilmingud. Haigus avaldub 6-15-aastaselt. Paroksüsme iseloomustab äkiline lihasnõrkuse areng, liikumatus öösel või hommikul, lihastoonuse langus, kõõluste refleksid, autonoomsed häired - pulsi labiilsus, vererõhk, liighigistamine. Rünnakud on osalised, hõlmates väikest rühma lihaseid ja üldistatud. Rünnaku ajal esinevad kardiovaskulaarse aktiivsuse rikkumised: süstoolne müra, EKG muutused. Teadvus on alati säilinud. Rünnaku keskmine kestus on mitu tundi, äärmiselt harva kestavad paroksüsmid mitu päeva. Rünnaku ajal on vere kaaliumisisaldus alla 2 mmol / l ja alla selle. Krambihoogude sagedus on erinev. Neid provotseerib süsivesikuterikka toidu ülesöömine, jahutamine, füüsiline aktiivsus.
Ravi. Kaaliumirikas dieet (ploomid, kuivatatud aprikoosid, kartul, rosinad). Rünnaku peatamiseks määratakse suukaudselt 10% kaaliumkloriidi lahus (1 supilusikatäis tunnis) või 0,5% lahus isotoonilises naatriumkloriidi lahuses intravenoosselt (2–2,5 g 500 ml lahuse kohta tunnis). Panangini on soovitatav kasutada ka intravenoosselt.
Paroksüsmaalse müolegiini hüperkaleemiline vorm (Gamstorpi tõbi). Seda haigust kirjeldas I. Gamstorp 1956. aastal. See pärineb autosoomselt dominantselt.
Kliinilised ilmingud. Haigus avaldub 1-5-aastaselt. Sümptomid on sarnased hüpokaleemilise vormi paroksüsmidele ja neid iseloomustab lihasnõrkuse äkiline areng, pleegia, lihastoonuse langus, kõõluste refleksid, autonoomsed häired. Erinevalt hüpokaleemilisest halvatusest areneb hüperkaleemiline halvatus tavaliselt päeva jooksul, sellega kaasnevad tugevad paresteesiad, see on kombineeritud näo lihaste, artikulatsiooniaparaadi nõrkusega ja on lühem (30-40 minutit). Rünnaku ajal tõuseb kaaliumisisaldus veres 6-7 mmol / l-ni. Rünnakute sagedus on erinev: päevast kuni mitme korrani kuus. Interiktaalsetel perioodidel neuroloogilised sümptomid puuduvad. Provotseerivad tegurid on nälgimine, füüsiline aktiivsus, väsimuse tekitamine.
Ravi. Kõrge süsivesikute, soola ja piiratud kaaliumisisaldusega toit. Sisestage intravenoosselt 40 ml 40% glükoosilahust koos insuliiniga subkutaanselt; 20 ml 10% kaltsiumkloriidi lahust intravenoosselt.
Normokaleemiline (perioodiline) halvatus. See on päritud autosomaalselt domineerival viisil.
Kliinilised ilmingud. Haigus avaldub enne 10. eluaastat. Selle eripäraks on suhteliselt aeglaselt (mõne päeva jooksul) paroksüsmaalselt süvenev mõõdukas nõrkus kehatüve lihastes, jäsemetes ja mälumislihastes, samuti sümptomite aeglane (1-2 nädalat) taandumine. Provotseerivad tegurid on pikaajaline uni, pikaajaline viibimine ühes asendis, hüpotermia.
Ravi. Soolarikas dieet. Määrake atsetasoolamiid (diakarb).
Voolu. Kõik paroksüsmaalse müopleegia vormid arenevad aeglaselt. Õigeaegse diagnoosi, erakorraliste meetmete ja diferentseeritud ravimteraapia prognoos on soodne.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos põhineb genealoogilisel analüüsil, kliinilise pildi omadustel, võttes arvesse haiguse alguse vanust, paroksüsmi esinemise aega (öösel, hommikul, päeval, määramata ajal ), lihasnõrkuse raskusaste, rünnaku sagedus ja kestus, provotseerivad tegurid, laboratoorsed andmed biokeemilised uuringud (lihaste bioelektrilise aktiivsuse sisaldus).
Haigust tuleks eristada primaarsete endokriinsete haiguste – türeotoksikoos, Conni tõbi (primaarne hüperaldosteronism), Addisoni tõbi jne – tagajärjel tekkivast müopleegiast.
Ravi. Kuvatakse lauasoolarikas dieet. Määra diakarb.
24.1.4. Müotoonia
Müotoonia on neuromuskulaarsete haiguste heterogeenne rühm, mida ühendab ühine iseloomulik lihastoonuse häirete kompleks, mis väljendub raskustes lihaste lõdvestamisel pärast aktiivset kontraktsiooni.
On pärilikud müotooniad (statsionaarsed aeglaselt progresseeruvad ja perioodilised, korduvad vormid) ja müotoonilised sündroomid.
Kaasasündinud müotoonia (Leiden-Thomseni tõbi). Seda haigust kirjeldas esmakordselt Leiden 1874. aastal. Thomsen juhtis 1876. aastal tähelepanu haiguse pärilikkusele, kasutades näitena oma perekonda (lapsed ja paljud sugulased – 20 tema pereliiget 4 põlvkonna jooksul kannatasid müotoonia all).
Sagedus 0,3-0,7 100 000 elaniku kohta. See on päritud autosomaalselt domineerival viisil. Meestel on läbitungimine suurem.
Patogenees. Olulised on rakumembraani läbilaskvuse rikkumised, ioonide ja vahendajate metabolismi muutused (kaltsium-troponiin-aktomüosiini lingi funktsionaalse suhte häired), kudede suurenenud tundlikkus atsetüülkoliini ja kaaliumi suhtes.
Patomorfoloogia. Valgusmikroskoopia näitab üksikute lihaskiudude hüpertroofiat; histokeemiliselt määratakse II tüüpi lihaskiudude suuruse vähenemine; elektronmikroskoopia näitab sarkoplasmaatilise retikulumi mõõdukat hüpertroofiat, mitokondrite kuju ja suuruse muutust ning müofibrillaarsete kiudude telofragma laienemist.
Kliinilised ilmingud. Esimest korda ilmnevad haiguse sümptomid peamiselt vanuses 8-15 aastat. Juhtivad tunnused on müotoonilised spasmid – raskused lihaste lõdvestamisel pärast aktiivset pinget. Müotoonilised spasmid paiknevad erinevates lihasrühmades, sagedamini käte, jalgade lihastes, mälumislihastes ja silma ringlihastes. Tugev sõrmede kokkusurumine, jalgade pikaajaline staatiline pinge, lõualuude sulgumine, silmade kissitamine põhjustavad toniseerivaid spasme. Lihaste lõdvestamise faas viibib pikka aega ja patsiendid ei saa kiiresti käsi lahti võtta, jalgade asendit muuta, suud ja silmi avada. Korduvad liigutused vähendavad müotoonilisi spasme. Lihaste mehaanilise erutatavuse suurenemine määratakse spetsiaalsete tehnikate abil: kui neuroloogiline haamer lööb esimese sõrme eminentsi, tuuakse see käele (mõnest sekundist minutini) - "pöidla sümptom", kui löökhaamer lööb vastu keelt, sellele tekib auk, ahenemine - "keele sümptom". Patsientide välimus on omapärane. Erinevate lihaste difuusse hüpertroofia tõttu meenutavad nad professionaalseid sportlasi. Palpatsioonil on lihased tihedad, kindlad, kuid objektiivselt on lihasjõud vähenenud. Kõõluste refleksid on normaalsed, rasketel juhtudel vähenenud.
Voolu. Haigus areneb aeglaselt. Tööalane konkurentsivõime säilib pikka aega.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos põhineb genealoogilisel analüüsil (autosoomne dominantne pärand), kliinilistel tunnustel (sportlik kehatüüp, difuusne lihashüpertroofia, müotooniline sündroom), globaalse elektromüograafia andmetel (müotooniline reaktsioon).
Seda haigust tuleks eristada teistest müotoonia vormidest, mõnikord progresseeruvate lihasdüstroofiate pseudohüpertroofsetest vormidest.
Ravi. Määrake difeniin (0,1-0,2 g 3 korda päevas 2-3 nädala jooksul), diakarb (0,125 g 2 korda päevas 2-3 nädala jooksul), kaltsiumipreparaadid (intravenoosne 10% kaltsiumkloriidi lahus 10 ml või kaltsiumglükonaat intramuskulaarselt). Eeldatakse, et difeniinil on pärssiv toime mono- ja polüsünaptilisele juhtivusele kesknärvisüsteemis ning diakarb muudab membraani läbilaskvust. Sobiv füsioteraapia galvaanilise krae ja kaltsiumiga lühikeste pükste näol, ravivõimlemine.
Düstroofiline müotoonia Rossolimo-Steinert-Kurshman. Seda haigust kirjeldas esmakordselt G. I. Rossolimo 1901. aastal ning seejärel Steinert ja Kurshman 1912. Sagedus on 2,5–5 juhtu 100 000 elaniku kohta. See on päritud autosomaalselt domineerival viisil.
Patogenees. Ebaselge. Eeldatakse membraanide esmast defekti.
Patomorfoloogia. Valgusmikroskoopia abil tuvastatakse atroofeerunud ja hüpertrofeerunud lihaskiudude kombinatsioon, sidekoe vohamine, lihaskoe asendamine rasv- ja sidekoega. Elektronmikroskoopiaga määratakse mitokondrite suuruse muutus, müofibrillaarse aparaadi hävimine ja sarkoplasmaatiline retikulum.
Kliinilised ilmingud. Esimesed haigusnähud ilmnevad 10-20-aastaselt. Iseloomulik on müotooniliste, müopaatiliste, neuroendokriinsete, kardiovaskulaarsete häirete kombinatsioon. Müotooniliste sümptomite kompleks, nagu ka Thomseni kaasasündinud müotoonia puhul, avaldub müotooniliste spasmide, suurenenud mehaanilise erutuvuse tõttu. Müotoonilise nähtuse raskus raske lihasdüstroofiaga haiguse hilisemates staadiumides nõrgeneb. Müopaatilisele sündroomile on iseloomulik lihaste patoloogiline väsimus, nõrkus, lihaste atroofia, mis paiknevad peamiselt näo-, kaela- ja distaalsete jäsemete lihastes. Atroofia tõttu on haigete välimus omapärane: pea on langetatud kaelale, nägu on miimiline, kõhn, eriti ajalises piirkonnas, silmalaud on poolrippunud, jalad ja käed on distaalsetes lõikudes kitsendatud. Tüüpilised "söödud" jalad, "ahvi" harjad. Peroneaalne kõnnak ("samm"), mõnikord koos "pardi" komponendiga proksimaalsete lihasrühmade atroofiaga. Lihastoonus väheneb, kõõluste refleksid kaovad varakult. Neuroendokriinsed häired on mitmekesised. Kõige silmatorkavamad muutused sugunäärmetes. Meestel täheldatakse sageli krüptorhidismi, libiido langust, impotentsust, naistel - menstruaaltsükli häireid. Paljudel patsientidel on varajane kiilaspäisus, hõrenemine ja naha kuivus. Kardiovaskulaarsed häired on püsivad. Esineb His-kimbu jalgade täielik või osaline blokaad, EKG madal pinge, arütmia.
Haigus areneb aeglaselt.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos tehakse genealoogilise analüüsi (autosoomne dominantne pärand), kliiniliste tunnuste (müotooniliste, müopaatiliste, neuroendokriinsete, kardiovaskulaarsete häirete kombinatsioon), globaalse elektromüograafia (müotooniline reaktsioon), biokeemiliste vereanalüüside (insuliiniresistentsus) põhjal.
Haigust tuleks eristada Thomseni kaasasündinud müotooniast, teistest müotoonilistest vormidest, progresseeruvatest lihasdüstroofiatest – distaalsest müopaatiast, neuraalsest amüotroofiast.
Ravi. Nagu kaasasündinud müotoonia puhul, annavad difeniin ja diakarb positiivse efekti. Näidatud on anaboolsete steroidide (retaboliil, nerobol, metüülandrostenediool) kasutamine. Dieet peaks vähendama kaaliumi kogust.
24.2. Püramidaalsed ja ekstrapüramidaalsed degeneratsioonid
24.2.1. Strümpeli perekondlik spastiline halvatus
Närvisüsteemi krooniline progresseeruv pärilik degeneratiivne haigus, mida iseloomustavad seljaaju külgmiste ja eesmiste nööride püramiidteede kahepoolsed kahjustused. A. Shtryumpel märkis 1866. aastal haiguse perekondlikku olemust. Kasutatakse ka nimetust "Erb-Charcot-Strumpeli perekondlik spastiline parapleegia".
Etioloogia ja patogenees. Haigus on pärilik, levib sagedamini autosoomselt dominantselt, harvem autosoom-retsessiivselt ja suguga seotud (X-kromosoom) tüüpi. Degeneratsiooni ja esmase biokeemilise defekti patogenees pole teada.
Patomorfoloogia. Kõige sagedamini on kahjustatud seljaaju nimme- ja rindkere osad, harvem ajutüvi. Külgmistes ja eesmistes nöörides, Gaulle'i kimpudes on püramiidsete radade sümmeetriline gliaaldegeneratsioon. Kirjeldatakse degeneratiivsete muutuste juhtumeid eesmise tsentraalse gyruse ajukoore, seljaaju eesmiste sarvede ja väikeajujuhtide rakkudes.
Kliinilised ilmingud. Haiguse areng toimub järk-järgult. Kõige sagedamini ilmnevad esimesed sümptomid teisel elukümnendil, kuigi haiguse alguse vanuses esineb suuri kõikumisi. Esialgu on jalgade jäikus ja kõndimisel väsimus, mis haiguse edenedes suureneb. Tekib iseloomulik spastiline kõnnak, ühinevad varus ja equinovarus deformatsioonid jalgadel, muutused Friedreichi labajala tüüpi jalgades, kõõluste ja lihaste kontraktuurid, eriti hüppeliigeses. Järk-järgult suureneb alajäsemete nõrkus, kuid alajäsemete täielikku halvatust ei täheldata. Patsientide kliiniline uurimine juba haiguse algstaadiumis näitab kõõluste reflekside suurenemist, painde- ja sirutajarühmade (Babinsky, Oppenheim, Rossolimo, Gordon, Schaeffer, Bekhterev-Mendel, Zhukovsky) varaseid patoloogilisi reflekse, jalgade klooneid. , põlvekaitsmed. Naha refleksid enamikul juhtudel säilivad, vaagnaelundite funktsioonid ei ole kahjustatud. Sensoorsed häired puuduvad. Intelligentsus on salvestatud. Palju hiljem on patoloogilises protsessis kaasatud ülemised jäsemed. Sageli ühinevad madalama spastilise parapareesiga nägemis- ja okulomotoorsete närvide kahjustuse sümptomid, nüstagm, düsartria, ataksia ja tahtlik värisemine.
Diagnoos ja diferentsiaaldiagnostika. Diagnoos ei tekita tavaliselt raskusi haiguse perekondliku olemuse ja tüüpilise kliinilise pildi olemasolul.
Ebatüüpilistel sporaadilistel juhtudel tuleks haigust eristada sclerosis multiplex'i spinaalsest vormist, amüotroofsest lateraalskleroosist, seljaaju kasvajatest ja muudest erinevate etioloogiatega patoloogilistest protsessidest, mis põhjustavad seljaaju kokkusurumist, samuti funikulaarsest müeloosist, neurosüüfilisest ja muudest vormidest. väikeaju-püramiidi degeneratsioonist. Sclerosis multiplex'i spinaalset vormi koos madalama spastilise parapareesiga iseloomustab taanduv kulg, üksikute sümptomite püsimatus ja ajutine pöörduvus, vaagnaelundite talitlushäired, kõhu reflekside kadumine või asümmeetria ja kahjustuse sümptomite asümmeetria üldiselt. , muutused vere ja tserebrospinaalvedeliku immunoloogilistes parameetrites. Otsustava tähtsusega on andmed haiguse päriliku olemuse kohta. Erinevalt amüotroofsest lateraalskleroosist algab Strümpeli tõbi noores eas, perifeerse motoorse neuroni kahjustuse tunnused puuduvad (fastsikulaarsed tõmblused, käe väikeste lihaste atroofia, iseloomulikud EMG muutused), bulbaarsed häired. Eristamisel ekstramedullaarsetest kasvajatest ja muu etioloogiaga seljaaju kompressiooni sündroomist, kasvajatele iseloomulikest segmentaalsetest sensoorsetest häiretest, jäsemete kahjustuste asümmeetriast, subarahnoidaalse ruumiploki olemasolust ja valgu-rakkude dissotsiatsioonist tserebrospinaalvedelikus lumbaalpunktsiooni ajal, on olulised. Neurosüüfilise puhul on erinevalt Strümpeli tõvest anamneesis märke naha ilmingutest. Kliinilises pildis on juhtivad seljaaju tagumiste nööride kahjustuste sümptomid, iseloomulikud pupillide häired, muutused veres, tserebrospinaalvedelikus.
Perekondliku spastilise parapleegia diferentsiaaldiagnostika koos teiste seljaaju degeneratiivsete kahjustustega on mõnikord keeruline. See aitab tuvastada teiste närvisüsteemi osade (väikeaju, okulaarne jne) kahjustuse sümptomeid.
Praegune ja prognoos. Haiguse kulg on aeglaselt progresseeruv; on pahaloomulisem kulg, kui see esineb varases eas. Haiguse hilise arenguga domineerivad hüpertensioon ja hüperrefleksia motoorsete häirete üle. Prognoos eluks on soodne. Puude aste sõltub närvisüsteemi düsfunktsiooni raskusastmest.
Ravi. Sümptomaatiline. Määrake lihastoonust vähendavad ravimid - midokalm, baklofeen, isoprotaan (skutamiil), rahustid: sibazon (seduxen), nozepaam (tazepaam), klotsepiid (elenium). Näidatud on füsioterapeutilised protseduurid, parafiinirakendused alajäsemete lihastele. Kasutatakse akupressuuri, refleksoloogiat, füsioteraapia harjutusi, vajadusel ortopeedilisi abinõusid. Näidatud on taastava ravi kursused: B-vitamiinid, metaboolsed ravimid: piratsetaam (nootropiil), püriditool (entsefabool), aminalon, tserebrolüsiin, aminohapped, ATP, kokarboksülaas, mikrotsirkulatsiooni parandavad ravimid.
24.2.2. Parkinsoni tõbi
Seda haigust kirjeldas esmakordselt inglise arst James Parkinson, kes nimetas seda värisemishalvatuseks. 1877. aastal lõpetas Jean Martin Charcot haiguse kliinilise kirjelduse. Haigus esineb 60-140 inimesel 100 000 elaniku kohta; selle sagedus suureneb järsult koos vanusega. Statistika järgi esineb värinahalvatus alla 60-aastastel elanikel 1% ja vanemas eas 5%. Mehed haigestuvad veidi sagedamini kui naised.
Etioloogia ja patogenees. Väriseva halvatuse ja parkinsonismi sündroomi kliinilised ilmingud tekivad närvisüsteemi ägedate ja krooniliste infektsioonide (Economo epideemiline entsefaliit, puuk-, viirus- ja muud tüüpi entsefaliit) tagajärjel. Haiguse põhjused võivad olla aju ateroskleroos, tserebrovaskulaarsed haigused, kasvajad, närvisüsteemi vigastused, pikaajaline fenotiasiiniravimite (kloorpromasiin, triftasiin), rauwolfia derivaadid, metüüldopa - ravimiparkinsonismi kasutamine. Parkinsonism võib areneda ägeda või kroonilise vingugaasi- ja mangaanmonooksiidi mürgistuse korral. Akineetilis-jäika sündroomi ilmnemisel võib olla oluline katehhoolamiinide metabolismi pärilik häire ajus või seda vahetust kontrollivate ensüümsüsteemide ebapiisavus. Haiguse perekondlikku olemust tuvastatakse sageli autosomaalse domineeriva päranditüübiga. Selliseid juhtumeid nimetatakse Parkinsoni tõveks. Erinevad eksogeensed ja endogeensed tegurid (ateroskleroos, infektsioonid, mürgistus, traumad) aitavad kaasa tõeliste defektide ilmnemisele katehhoolamiinide metabolismi mehhanismides subkortikaalsetes tuumades ja haiguse algusele.
Treemori halvatuse ja parkinsonismi sündroomi peamine patogeneetiline seos on katehhoolamiinide (dopamiini, norepinefriini) vahetuse rikkumine ekstrapüramidaalsüsteemis. Dopamiin täidab motoorsete tegude rakendamisel sõltumatut vahendaja funktsiooni. Tavaliselt on dopamiini kontsentratsioon basaalganglionides kordades suurem kui selle sisaldus teistes närvisüsteemi struktuurides. Atsetüülkoliin on ergastav vahendaja juttkeha, globus pallidus'e ja substantia nigra vahel. Dopamiin on selle antagonist, toimides inhibeerivalt. Substantia nigra ja globus palliduse lüüasaamisega väheneb dopamiini tase sabatuumas ja putamenis, dopamiini ja noradrenaliini suhe on häiritud ning ekstrapüramidaalsüsteemi funktsioonide häired. Tavaliselt moduleeritakse impulss sabatuuma, putameni, mustandi ja globus palliduse stimuleerimise suunas. Kui substantia nigra funktsioon on välja lülitatud, blokeeritakse impulsid, mis tulevad ajukoore ekstrapüramidaalsetest tsoonidest ja juttkehast seljaaju eesmistesse sarvedesse. Samal ajal saavad eesmiste sarvede rakud patoloogilisi impulsse globus palliduselt ja mustandilt. Selle tulemusena suureneb impulsside tsirkulatsioon seljaaju alfa- ja gamma-motoorsete neuronite süsteemis, kus ülekaalus on alfa aktiivsus, mis põhjustab lihaskiudude pallidar-nigraalse jäikuse ja treemori - peamised parkinsonismi tunnused. .
Patomorfoloogia. Parkinsonismi peamisi patoanatoomilisi muutusi täheldatakse substantia nigras ja globus palliduses degeneratiivsete muutuste ja närvirakkude surma kujul. Surnud rakkude asemele tekivad gliaalelementide kasvukolded või jäävad tühimikud.
Kliinilised ilmingud. Peamine kliiniline sündroom on akineetilis-jäik või hüpertensiivne-hüpokineetiline. Hüpo- ja akineesia on iseloomulikud värisevatele halvatustele ja parkinsonismile. Ilmub omapärane painutusasend: pea ja torso on ette kallutatud, käed küünarnukist poolkõverdatud, randme- ja falangeaalliigesed, sageli tihedalt rindkere, torso külgpindadele kõverdatud, jalad poolkõverdatud. põlveliigesed. Märgitakse näoilmete vaesust. Vabatahtlike liigutuste kiirus aeglustub haiguse arenedes järk-järgult, mõnikord võib üsna varakult tekkida täielik liikumatus. Kõnnakut iseloomustavad väikesed segavad sammud. Sageli on kalduvus tahtmatule edasijooksule (tõukejõule). Kui patsienti edasi lükata, jookseb ta nii, et ta ei kukuks, justkui "jõudes oma raskuskeskmele järele". Sageli viib tõuge rinnale tagasijooksu (retropulsioon), küljele (lateropulsioon). Neid liigutusi täheldatakse ka siis, kui proovite istuda, püsti tõusta, pea tagasi visata. Sageli, väljendunud sündroomiga, sarnanevad patsiendi poosid kataleptilistele. Akineesi ja plastiline hüpertensioon on eriti väljendunud näolihastes, mälumis- ja kuklalihastes ning jäsemete lihastes. Kõndimisel ei esine käte sõbralikke liigutusi (ahheirokinees). Kõne on vaikne, monotoonne, ilma modulatsioonideta, fraasi lõpus kipub tuhmuma.
Jäseme passiivse liikumise korral täheldatakse antagonistlike lihaste toonuse suurenemise tõttu teatud tüüpi lihaste vastupanu, "hammasratta" nähtust (jääb mulje, et liigendpind koosneb kahe hammasratta sidurist) . Antagonisti lihaste toonuse tõusu passiivsete liigutuste ajal saab määrata järgmise meetodiga: kui tõstate lamava inimese pea ja seejärel järsult vabastate käe, ei kuku pea padjale, vaid kukub suhteliselt. sujuvalt. Mõnikord on pea lamavas asendis mõnevõrra üles tõstetud - "kujuteldava padja" nähtus.
Treemor on Parkinsoni sündroomi iseloomulik, kuigi mitte kohustuslik sümptom. See on jäsemete, näolihaste, pea, alalõua, keele rütmiline, regulaarne, tahtmatu värisemine, puhkeolekus rohkem väljendunud, väheneb aktiivsete liigutustega. Võnkesagedus 4-8 sekundis. Mõnikord märgitakse sõrmeliigutusi "pillide veeremise", "müntide lugemise" kujul. Treemor suureneb erutusega, praktiliselt kaob une ajal.
Vaimsed häired väljenduvad algatusvõime, aktiivsuse kadumises, silmaringi ja huvide ahenemises, erinevate emotsionaalsete reaktsioonide ja afektide järsu langusena, aga ka mõtlemise mõningase pinna- ja aeglustumisena (bradüfreenia). Täheldatakse bradüpsühhiat - raske aktiivne üleminek ühelt mõttelt teisele, akairiya - kleepuvus, viskoossus, egotsentrism. Mõnikord esineb vaimse erutuse paroksüsme.
Vegetatiivsed häired avalduvad näo- ja peanaha rasvasuse, seborröa, hüpersalivatsiooni, liighigistamise, distaalsete jäsemete troofiliste häiretena. Selgub posturaalsete reflekside rikkumine. Mõnikord on spetsiaalsete uurimismeetoditega hingamine ebaregulaarne sageduse ja sügavuse poolest. Kõõluste refleksid reeglina ilma kõrvalekalleteta. Aterosklerootilise ja postentsefaliitilise parkinsonismiga saab määrata kõõluste reflekside suurenemist ja muid püramidaalse puudulikkuse tunnuseid. Postentsefaliitilise parkinsonismiga tekivad niinimetatud okulogüürilised kriisid – pilk fikseeritakse mitmeks minutiks või tunniks ülespoole; vahel visatakse pea taha. Kriise saab kombineerida lähenemise ja majutuse rikkumisega (progresseeruv supranukleaarne halvatus).
On tavaks eristada väriseva halvatuse ja parkinsonismi mitmeid kliinilisi vorme; jäik-bradükineetiline, värin-jäik ja värisev. Jäik-bradükineetilist vormi iseloomustab lihastoonuse tõus vastavalt plastilisele tüübile, aktiivsete liigutuste progresseeruv aeglustumine kuni liikumatuseni; ilmnevad lihaste kontraktuurid, patsientide painutaja asend. Seda parkinsonismi vormi, mis on selle käigus kõige ebasoodsam, täheldatakse sagedamini aterosklerootilise ja harvemini postentsefaliidi parkinsonismi korral. Tremor-jäika vormi iseloomustab jäsemete, peamiselt nende distaalsete osade treemor, millele haiguse arenedes lisandub ka vabatahtlike liigutuste jäikus. Parkinsonismi värisevat vormi iseloomustab jäsemete, keele, pea ja alalõua keskmise ja suure amplituudiga pidev või peaaegu konstantne treemor. Lihastoonus on normaalne või veidi tõusnud. Vabatahtlike liigutuste tempo on salvestatud. Seda vormi esineb sagedamini postentsefaliidi ja traumajärgse parkinsonismi korral.
Laboratoorsete ja funktsionaalsete uuringute andmed. Traumajärgse parkinsonismi korral tuvastatakse tserebrospinaalvedeliku rõhu tõus selle normaalse rakulise ja valgu koostisega. Süsinikmonooksiidi mürgistusest põhjustatud parkinsonismi korral leitakse veres karboksühemoglobiini, mangaani parkinsonismi korral - mangaani jälgi veres, uriinis ja tserebrospinaalvedelikus. Globaalne elektromüograafia värisemise halvatuse ja parkinsonismi korral näitab lihaste elektrogeneesi rikkumist - lihaste bioelektrilise aktiivsuse suurenemist puhkeolekus ja potentsiaalide rütmiliste rühmade tühjenemise olemasolu. Elektroentsefalograafia näitab aju bioelektrilises aktiivsuses valdavalt hajusaid mitte-jämedaid muutusi.
Diagnoos ja diferentsiaaldiagnostika. Esiteks tuleks Parkinsoni tõbe eristada Parkinsoni sündroomist. Okulomotoorsed sümptomid on iseloomulikud postentsefaliidile parkinsonismile; võib täheldada torticollist, torsioondüstoonia nähtusi, mida kunagi ei täheldata väriseva halvatusega. Esinevad unehäired, hingamisteede düskineesiad koos haigutamishoogudega, köha, rasvkoe häired, autonoomsed paroksüsmid. Traumajärgset parkinsonismi saab usaldusväärselt diagnoosida noortel ja keskealistel patsientidel. Haigus areneb pärast rasket, mõnikord korduvat traumaatilist ajukahjustust. Posttraumaatilise parkinsonismi korral ei ole iseloomulikud anteretropulsia, pilgukrambid, närimis-, neelamis-, hingamishäired ja kataleptoidsed nähtused. Samal ajal on sagedased vestibulaarsed häired, intelligentsuse ja mälu halvenemine, nägemishallutsinatsioonid (ajukoore kahjustuse tõttu). Sageli toimub patoloogilise protsessi regeneratiivne kulg või stabiliseerumine. Mangaani parkinsonismi diagnoosimisel on oluline anamnees (teave töö kohta kokkupuutel mangaani või selle oksiididega), mangaani tuvastamine bioloogilistes vedelikes. Oxycarbon parkinsonismi diagnoos põhineb karboksühemoglobiini määramisel veres.
Aterosklerootilise parkinsonismi korral on värisemine ja jäikus kombineeritud aju ateroskleroosi tunnustega või tekivad pärast ägedaid tserebrovaskulaarseid õnnetusi. Fokaalsed neuroloogilised sümptomid ilmnevad püramidaalse puudulikkuse, väljendunud pseudobulbaarsete sümptomite kujul. Sageli tuvastatakse ühepoolne jäikus ja jäikus. Verest leitakse ateroskleroosile iseloomulik düslipideemia. Teatud muutused REG-is registreeritakse pulsilainete lamenemise kujul.
Parkinsoni tõbe meenutavat kliinilist pilti võib täheldada seniilse aterosklerootilise dementsuse korral, mida iseloomustavad enim rasked psüühikahäired kuni dementsuseni. Jäikus ja jäikus on mõõdukad, treemor tavaliselt puudub. Eraldi parkinsonismi kliinilisi ilminguid võib leida teiste pärilike degeneratiivsete närvisüsteemi haiguste puhul: Friedreichi ataksia, olivopontotserebellaarne atroofia, ortostaatiline hüpokineesia, Creutzfeldt-Jakobi tõbi. Nende haiguste puhul on koos akineetilise-jäika sümptomitega ka progresseeruv väikeaju ataksia.
Praegune ja prognoos. Haigus areneb pidevalt. Erandiks on mõned uimastimürgitusest põhjustatud vormid (ravimite ärajätmisel võib seisund paraneda). On üldtunnustatud, et ravi algstaadiumis võib vähendada sümptomite raskust, aeglustada haiguse progresseerumist. Hilisematel etappidel on terapeutilised meetmed vähem tõhusad. Haigus põhjustab mõne aasta jooksul puude. Isegi ravi levodopaga aeglustab praegu kulgu lühikeseks ajaks. See kinnitab seisukohta, et haigus ei põhine ainult esmasel biokeemilisel defektil, vaid ka seni uurimata neuropatoloogilisel protsessil.
Ravi. Väriseva halvatuse ja parkinsonismi sündroomiga patsientide ravi peaks olema terviklik, pikaajaline ja hõlmama spetsiifilisi parkinsonismivastaseid ravimeid, rahusteid, füsioteraapiat, füsioteraapiat, psühhoteraapiat, võttes arvesse etioloogilist tegurit, patsientide vanust, kliinilist vormi ja haiguse staadiumi, samuti kaasuvate haiguste esinemine. Kergete vormide korral määratakse esmalt amantadiin (midantan) ja parasümpatolüütikumid, kuna need põhjustavad vähem kõrvaltoimeid. Kandke tsentraalseid parasümpatolüütikume (tsüklodool, narkopaan), püridoksiini, amantadiini, dopamiini retseptori agoniste (bromokriptiin, lisuriid).
Parkinsonismi raskete kliiniliste ilmingute korral on praegu põhiravim levodopa, tavaliselt kombinatsioonis dekarboksülaasi inhibiitoriga. Annuseid suurendatakse aeglaselt, mitme nädala jooksul, kuni saavutatakse kliiniline toime. Ravimi kõrvaltoimed on düstoonilised häired ja psühhoosid. Levodopa, sattudes kesknärvisüsteemi, dekarboksüleeritakse dopamiiniks, mis on vajalik basaalganglionide normaalseks talitluseks. Ravim mõjutab peamiselt akineesiat ja vähemal määral ka muid sümptomeid. Levodopa kombineerimisel dekarboksülaasi inhibiitoriga on võimalik levodopa annust vähendada ja seeläbi kõrvaltoimete riski.
Sümptomaatiliste parkinsonismivastaste ravimite arsenalis on suurel kohal antikolinergilised ravimid, mis m- ja n-kolinergiliste retseptorite blokeerimisega aitavad lõdvestada vööt- ja silelihaseid, vähendada vägivaldseid liigutusi ja bradükineesiat. Need on looduslikud ja sünteetilised atropiinilaadsed ravimid: bellazon (romparkiin), norakiin, kombipark. Kasutatakse ka fenotiasiini preparaate: dinezin, deparcol, parsidol, diprasine. Parkinsonismi raviks kasutatavate ravimite mitmekesisuse peamiseks põhjuseks on nende terapeutilise efektiivsuse puudumine, kõrvaltoimete esinemine, individuaalne talumatus ja kiire sõltuvus nendest.
Kirurgia. Vaatamata parkinsonismi meditsiinilises ravis saavutatud suurtele edusammudele on selle võimalused mõnel juhul piiratud.
Kõige laialdasemalt kasutatav ravim levodopa leevendab tõhusamalt haiguse sümptomeid, nagu akineesia, üldine jäikus, vähemal määral mõjutab see lihaste jäikust ja värinat. Umbes 25% patsientidest on see ravim praktiliselt ebaefektiivne või halvasti talutav.
Nendel juhtudel on näidustused subkortikaalsete sõlmede stereotaksilise operatsiooni jaoks. Tavaliselt tehakse talamuse ventrolateraalse tuuma, subtalamuse struktuuride või globus palliduse lokaalne hävitamine.
Operatsiooni abil on enamikul juhtudel võimalik saavutada positiivne efekt – lihastoonuse langus, treemori nõrgenemine või lakkamine ning hüpokineesia vähenemine.
Operatsioon tehakse tavaliselt parkinsonismi sümptomitega võrreldes vastupidisel küljel. Näidustuste korral viiakse läbi subkortikaalsete struktuuride kahepoolne hävitamine.
Viimastel aastatel on parkinsonismi raviks kasutatud ka loote neerupealiste koe siirdamist juttkehasse. Selliste operatsioonide kliinilisest efektiivsusest on veel vara rääkida.
Subkortikaalsete struktuuride stereotaktilisi operatsioone kasutatakse ka teiste haiguste korral, millega kaasnevad vägivaldsed liigutused (hemiballism, koreoatetoos, tortikollis ja mõned teised).
Parkinsoni tõve ja parkinsonismi tööalane konkurentsivõime sõltub motoorsete häirete raskusastmest ja kutsetegevuse tüübist. Kergete ja mõõdukate motoorsete funktsioonide häiretega jäävad patsiendid pikaks ajaks töövõimeliseks erinevat tüüpi vaimse töö, aga ka füüsilise pingega mitteseotud töö ning täpsete ja koordineeritud liigutuste tegemisega. Haiguse raskete ilmingute korral ei saa patsiendid töötada ja vajavad kõrvalist abi.
24.2.3. Hepatotserebraalne düstroofia
Hepatotserebraalne düstroofia (hepatolentikulaarne degeneratsioon, Westphal-Wilson-Konovalovi tõbi) on krooniline progresseeruv pärilik degeneratiivne haigus, mida iseloomustab kesknärvisüsteemi subkortikaalsete sõlmede ja maksa kombineeritud kahjustus. 1883. aastal kirjeldas K. Westphal ja 1912. aastal S. Wilson. Mõiste "hepatotserebraalne düstroofia" pakkus välja N.V. Konovalov.
Etioloogia ja patogenees. Haigus on pärilik. Pärandi tüüp on autosoomne retsessiivne. Juhtiv patogeneetiline seos on geneetiliselt määratud tseruloplasmiini valgu sünteesi rikkumine, mis on osa vaske transportivast a2-globuliinist. Selle tulemusena tekib veres kõrge vase kontsentratsioon ja see ladestub elunditesse ja kudedesse, peamiselt maksas, ajus, sarvkestas, aga ka neerudes ja teistes elundites. Vase toksiline toime on seotud oksüdatiivsete ensüümide sulfhüdrüülrühmade blokeerimisega, mis põhjustab raku redoksprotsesside katkemist.
Patomorfoloogia. Ajus, maksas, neerudes, põrnas, sarvkestas, iirises, silmaläätses määratakse degeneratiivsed muutused, mis on kõige enam väljendunud subkortikaalsetes tuumades. Samuti on närvirakkudes düstroofsed muutused, ajukoe fokaalne pehmenemine koos mikrotsüstide moodustumisega ja glia kasv. Ilmuvad muutused ajukoe väikestes veresoontes, nende ümber esinevad hemorraagiad, perivaskulaarne turse. Maksatsirroos on haiguse püsiv sümptom.
Kliinilised ilmingud. Need koosnevad kesknärvisüsteemi ja siseorganite kahjustuse sümptomitest. Patsientidel ilmnevad ja suurenevad lihaste jäikus, mitmesugused hüperkineesiad, pseudobulbaarsed sümptomid, intelligentsuse progresseeruv langus, maksafunktsiooni kahjustus ja vikerkesta muutus. Juhtiv on ekstrapüramidaalsete häirete sündroom: kehatüve, jäsemete, näo, neelu lihaste jäikus ja sellest tulenevalt kõnnaku-, neelamis- ja kõnehäired. Paralleelselt tekib erineva iseloomuga hüperkinees: treemor, atetoos, torsioondüstoonia, tahtlik värisemine, mis suurenevad tahtlike liigutuste sooritamisel. Hüperkineesiad on mitterütmilised.
Sõltuvalt kliiniliste ilmingute raskusest ja kombinatsioonist, haiguse ilmnemise vanusest ja maksakahjustuse astmest eristatakse nelja hepatotserebraalse düstroofia vormi.
1. Varajane jäik-arütmohüperkineetiline vorm on kõige pahaloomulisema kuluga. Neuroloogilised ilmingud arenevad vanuses 7-15 aastat. Tavaliselt eelnevad sellele maksakahjustuse nähud. Kliinilises pildis domineerivad lihaste jäikus ja hüperkinees.
2. Värisevad-jäigad ja värisevad vormid, mis avalduvad hilisemas eas (17-20 aastat). Iseloomustab samaaegne jäikuse ja värisemise ilmnemine, mis on sageli haiguse esimene märk; järk-järgult kasvades võib see muutuda üldiseks, haarates kehatüve, jäsemete, näo, lõualuude, pehme suulae, epiglottise, häälepaelte, hingamislihaste, diafragma lihaseid. Neelamine on häiritud, kõne muutub skandaalseks. Sageli täheldatakse olulisi vaimseid muutusi.
3. Ekstrapüramidaal-kortikaalne vorm, isoleeritud N.V. Konovalovile on iseloomulik kõrgemate ajufunktsioonide häire, halvatuse esinemine, sageli epilepsiahood, intelligentsuse jäme langus koos isiksuse muutustega.
4. Kõhuvormi iseloomustab valdav maksafunktsiooni rikkumine. Neuroloogilised sümptomid ühinevad haiguse hilisemates staadiumides.
Praegune ja prognoos. Kursus edeneb pidevalt. Oodatav eluiga sõltub haiguse kliinilisest vormist, alustatud ravi õigeaegsusest. Ravita patsientide keskmine eluiga on umbes 6 aastat.
Laboratoorsete ja funktsionaalsete uuringute andmed. Tseruloplasmiini sisalduse oluline vähenemine vereseerumis (alla 10 U kiirusega 25-45 U), hüpoproteineemia, hüperkupuria (kuni 1000 mcg / päevas ja rohkem kiirusega 150 mcg / päevas) ja hüperaminoatsiduuria (kuni 1000 mg / päevas kiirusega 350 mg). / päevas). Samuti võib esineda ammoniaagi sisalduse suurenemine veres, muutused maksaanalüüsides. Hepatotserebraalse düstroofia patognoomiline tunnus on Kaiser-Fleischeri sarvkesta rõngas, mis on vaske sisaldava pigmendi ladestumine piki vikerkesta perifeeriat.
diferentsiaaldiagnostika. Haigust tuleks eristada letargilisest entsefaliidist, koreast, degeneratiivsetest subkortikaalsetest haigustest, hulgiskleroosist. Perekonna ajaloo tuvastamine, iseloomulik kliiniline pilt, Kaiser-Fleischeri sarvkesta rõngas, madal tseruloplasmiini tase veres ja vase eritumise suurenemine uriiniga patsientidel ja nende sugulastel võimaldavad diagnoosida hepatotserebraalset düstroofiat.
Ravi. Ravi peamine eesmärk on eemaldada kehast liigne vask. Selleks kasutatakse tioolpreparaate, mille hulka kuuluvad unitiool, dekaptool ja D-penitsillamiin. Annused valitakse individuaalselt. 0-penitsillamiin määratakse keskmiselt annuses 0,45–2 g päevas pärast sööki. Ravimit tuleb võtta kogu elu.
Ravi on kõige tõhusam haiguse varases staadiumis. Unitiol määratakse korduvate kursustena 5 ml 5% lahust intramuskulaarselt iga päev või ülepäeviti (25 süstist koosneva kuuri jaoks 5-6-kuulise pausiga). Ravi kombineeritakse maksafunktsiooni parandavate ravimitega. Soovitatav on eridieet, piirates vaske (maks, seened, šokolaad, austrid jne), loomseid rasvu ja valke sisaldavaid toite. Toit peaks olema rikas vitamiinide ja süsivesikute poolest.
24.2.4. Torsiooni düstoopia
Närvisüsteemi krooniline progresseeruv haigus, mis kliiniliselt väljendub lihastoonuse muutustes ning kehatüve ja jäsemete lihaste tahtmatutes toonilistes kontraktsioonides.
Etioloogia ja patogenees. Esineb idiopaatiline (perekondlik) torsioon ja sümptomaatiline düstoonia. Idiopaatilise torsioondüstoonia puhul on pärilikkuse tüüp nii autosoomne domineeriv kui ka autosoomne retsessiivne. Sümptomaatiline torsioondüstoonia esineb hepatotserebraalse düstroofia, Huntingtoni korea, ajukasvajate, epideemilise entsefaliidi, tserebraalparalüüsi korral. On märke, et päriliku torsioondüstoonia patogeneesis on oluline dopamiini metabolismi rikkumine. Nende patsientide uurimine näitas dopamiin-β-hüdroksülaasi sisalduse suurenemist vereseerumis.
Patomorfoloogia. Düstroofilisi muutusi leitakse peamiselt väikestes neuronites läätsekujulise tuuma kesta piirkonnas, harvemini teistes basaalganglionides.
Kliinilised ilmingud. Haigus areneb järk-järgult, 2/3 juhtudest alla 15-aastastel. Lapsepõlves võivad haiguse esimesed sümptomid olla kõnnihäired, spastiline tortikollis; täiskasvanutel on esmased generaliseerunud vormid sagedasemad. Sünergistlike ja antagonistlike lihaste funktsioonide vahekorra rikkumise tulemusena tekivad kehatüve, pea, vaagnavöötme ja jäsemete lihaste vägivaldsed pikaajalised toonilised kokkutõmbed, tavaliselt pöörleva iseloomuga koos ateoidsete liigutustega. sõrmedes. Tundub, et lihased tõmbuvad pidevalt kokku, et antagonistide toimest üle saada. Tekkivad poosid, isegi kõige ebamugavamad, püsivad pikka aega. Hüperkineesiad süvenevad erutusest, aktiivsetest liigutustest ja kaovad une ajal. Aegamööda, haiguse progresseerumisel, muutub patsiendi kehahoiak püsivalt düstoonseks, suureneb nimmepiirkonna lordoos, puusa paindumine ning käte ja jalgade mediaalne pöörlemine. Sõltuvalt düstooniliste nähtuste levimusest eristatakse haiguse lokaalseid ja üldistatud vorme. Lokaalsete düstooniliste sümptomitega tekib üksikute lihasrühmade tooniline kokkutõmbumine, tahtlikud liigutused on häiritud ja kehahoiak on ebanormaalne. Selliste sümptomite hulka kuuluvad spastiline tortikollis, kirjude spasm, oromandibulaarne düstoonia (suu avanemine ja sulgumine ning keele tahtmatud liigutused), blefarospasm, buccofacial, bukaalne-lingvaalne düstoonia, koreoatetoos.
Praegune ja prognoos. Enamikul juhtudel areneb haigus pidevalt. Mõnikord on remissiooni kestus erinev. Kiiresti tekib patsientidel sügav puue ja surm, eriti üldistatud kujul.
Ravi. Pikaajaline, sümptomaatiline. Kasutatakse antikolinergiliste ja rahustite kombinatsioone, mõnel juhul on levodopa kasutamine efektiivne. Samuti on ette nähtud haloperidool või reserpiin. Väga harva kasutavad subkortikaalsete tuumade stereotaksilisi operatsioone.
24.2.5. Huntingtoni korea
Krooniline progresseeruv pärilik degeneratiivne haigus, mida iseloomustab suurenev koreiline hüperkineesia ja dementsus. Kirjeldas J. Huntington 1872. aastal. Huntingtoni korea esinemissagedus on 2–7 juhtu 100 000 elaniku kohta. Kasutatakse ka terminit "koreiline dementsus".
Etioloogia ja patogenees. Haigus on pärilik. Pärandiviis on autosoomne dominantne kõrge penetrantsusega (80-85%). Mehed haigestuvad sagedamini. Patogenees pole hästi mõistetav. Mõnel juhul leiti ajurakkudes GABA puudust, musta aine rakkudes rauasisalduse suurenemist ja dopamiini metabolismi rikkumisi. Liikumishäirete patofüsioloogilist mehhanismi on uuritud. Strionigraalsete ühenduste blokeerimine põhjustab kontrolli puudumist liigutuste samaaegse ja lihastoonuse üle substantia nigrast, mis edastab premotoorsest ajukoorest saadud impulsid ebakorrapärases järjestuses seljaaju eesmiste sarvede rakkudesse.
Patomorfoloogia. Leitakse aju atroofia. Subkortikaalsetes ganglionides, peamiselt putamenis ja sabatuumas, määratakse väikeste ja suurte rakkude degeneratiivsed muutused, nende arvu vähenemine ja gliiaelementide kasv.
Kliinilised ilmingud. Tavaliselt tekib haigus 30-aastaselt ja vanemad. Esimesed sümptomid võivad olla intellektihäired, hiljem areneb järk-järgult dementsus. Samal ajal ilmneb koreiline hüperkinees: kiired, ebaregulaarsed, ebaühtlased liigutused erinevates lihasrühmades. Tahtlike liigutuste sooritamine on hüperkineesi tõttu raskendatud ja sellega kaasnevad mitmed mittevajalikud liigutused. Näiteks kõndides teevad patsiendid grimassi, žestikuleerivad, kükitavad ja sirutavad käed laiali. Kuid isegi raske hüperkineesi korral, eriti haiguse alguses, võivad nad seda teadlikult alla suruda. Kõne on raske ja sellega kaasnevad ka liigsed liigutused. Lihastoonus väheneb. Jäsemete pareesi ja muid fokaalseid neuroloogilisi sümptomeid ei määrata. Sageli täheldatakse endokriinseid ja neurotroofilisi häireid. 5-16% juhtudest diagnoositakse Huntingtoni korea ebatüüpiline akineetilis-jäik variant. Samal ajal areneb akineetilis-jäik sündroom koos progresseeruva intellektuaalse degradatsiooni ja mõõdukalt väljendunud koreilise hüperkineesiga. Vägivaldsetest liigutustest domineerib koreoatetoos.
Voolu. Haigus areneb pidevalt. Selle kestus on 5-10 aastat alates esimeste sümptomite ilmnemisest. Healoomulisemat kulgu täheldatakse ebatüüpilise akineetilis-jäika vormiga.
Laboratoorsete ja funktsionaalsete uuringute andmed. EEG näitab hajusaid muutusi aju bioelektrilises aktiivsuses. Kompuutertomograafia ja MRT abil tuvastatakse vatsakeste laienemine ja nn talamuse depressioon, kui haigus on seotud selle väikeste rakkude kahjustusega, leitakse ajukoore atroofia tunnuseid. Vere lümfotsüütide röntgenkiirguse tundlikkuse uuringu põhjal on viiteid haiguse varajase prekliinilise diagnoosimise võimalusele.
Diagnoos ja diferentsiaaldiagnostika. Huntingtoni korea ebatüüpilistel juhtudel võib diagnoosimine olla keeruline.
Kõikidel juhtudel on suure tähtsusega haiguse perekondlik iseloom, ajukahjustuse muude fokaalsete sümptomite tuvastamine, haiguse kulgemise iseloom, muutused tserebrospinaalvedelikus ja muud diagnostilised kriteeriumid.
Huntingtoni korea eristamine tuleneb ajukasvajate korral esinevast koreaalsest sündroomist, süüfilisest, entsefaliidist, vaskulaarhaigustest, aga ka seniilsest (seniilsest) koreast.
Ravi. Hüperkineesi pärssimiseks on ette nähtud dopamiini antagonistid. Need on fenotiasiini seeria ravimid - triftasiin (7,5-10 mg päevas) kombinatsioonis rahustite, dopegiti, reserpiiniga. Katsed ravida Huntingtoni korea all kannatavaid patsiente stereotaksiliste operatsioonidega ebaõnnestusid.
24.2.6. Friedreichi haigus
Friedreichi perekondlik ataksia on pärilik degeneratiivne närvisüsteemi haigus, mida iseloomustab seljaaju tagumise ja külgmiste nööride kahjustuse sündroom. Pärandi tüüp on autosoomne retsessiivne, patoloogilise geeni mittetäieliku läbitungimisega. Mehed ja naised haigestuvad võrdselt sageli.
Patomorfoloogia. Degeneratiivseid muutusi leitakse seljaaju tagumiste ja külgmiste nööride radades, peamiselt Gaulli kimpudes, vähemal määral - Burdakh, Flexig, Gowers, püramiidtrakti kiud, tagumised juured, aga ka seljaaju rakkudes. väikeajukoor, subkortikaalsed ganglionid ja ajukoor.
Kliinilised ilmingud. Haiguse algus viitab 6-15 eluaastale. Haiguse esimene sümptom on ebakindel kõnnak, mida Charcot iseloomustas kui tabetilist väikeaju. Algstaadiumis väljendub ataksia peamiselt jalgades. Haiguse progresseerumisel levivad koordinatsioonihäired ülemistele jäsemetele ja näole. Neuroloogilisel uuringul tuvastatakse ulatuslik nüstagm, ataksia kätes ja jalgades, adiadohokinees, düsmetria, segane kõne, lihas-liigese tundlikkuse häired ja vibratsioonitundlikkus. Käekiri muutub. Varajane sümptom on kõõluste ja perioste reflekside vähenemine ja seejärel väljasuremine. Lihastoonus väheneb. Haiguse hilisemates staadiumides ei ole haruldased ala- ja seejärel ülemiste jäsemete aferentsed pareesid, patoloogilised püramiidrefleksid ja distaalne lihaste atroofia. Intelligentsus väheneb.
Haigus areneb aeglaselt. Keskmine eluiga on 10-15 aastat selle väljatöötamise hetkest.
Diagnoos ja diferentsiaaldiagnostika. Haigust tuvastatakse iseloomulike sümptomite põhjal - Friedreichi jalatüüpi labajala deformatsioonid (kõrge kaar, varvaste põhifalangide pikenemine ja lõppfalangide paindumine), müokardi kahjustus, endokriinsed häired.
Seda haigust tuleks eristada aju süüfilisest, hulgiskleroosist, funikulaarsest müeloosist ja muudest väikeaju degeneratsiooni vormidest.
Ravi. Kasutatakse sümptomaatilisi vahendeid: tugevdavad ravimid, füsioteraapia harjutused, massaaž. Mõnel juhul viiakse läbi jala deformatsiooni kirurgiline korrigeerimine.
24.2.7. Pierre Marie pärilik väikeaju ataksia
Pierre Marie väikeaju ataksia on pärilik degeneratiivne haigus, millega kaasneb väikeaju ja selle radade esmane kahjustus. Pärandi tüüp on autosoomne dominantne. Haigus esineb 20-aastastel ja vanematel inimestel.
Patomorfoloogia. Selgub ajukoore rakkude ja väikeaju tuumade degeneratiivne kahjustus, seljaaju külgmistes funikulites paiknevad spinotserebellaarsed rajad, silla ja pikliku medulla tuumades.
Kliinilised ilmingud. Haigus väljendub väikeaju ja selle ühenduste talitlushäiretes. Ataksiat täheldatakse koordineerivate testide sooritamisel, kõnnihäireid, skandeeritud kõnet, tahtlikku värisemist, nüstagmi. Väikeaju sümptomid on kombineeritud mõõdukate või raskete püramidaalse puudulikkuse tunnustega (kõõluste ja luuümbrise reflekside suurenemine, jala kloonus) ja mõnikord ka okulomotoorsete häiretega (strabismus, ptoos, konvergentsi puudulikkus). Iseloomulik tunnus on intelligentsuse väljendunud langus erineval määral.
Diagnoos ja diferentsiaaldiagnostika. Suurimad raskused tekivad Pierre Marie ja Friedreichi ataksia päriliku väikeaju ataksia eristamisel. Arvesse tuleb võtta haiguse pärilikkuse tüüpi, esimeste sümptomite ilmnemise vanust, kõõluste reflekside muutuse olemust (Friedreichi ataksia korral need vähenevad), nägemis- ja okulomotoorsete häirete esinemist. Pierre Marie ataksia, jalalaba ja luustiku deformatsioonid. Erinevalt Pierre Marie perekondlikust ataksiast iseloomustab hulgiskleroosi taanduv kulg, madalama spastilise parapareesi raskusaste ja vaagnaelundite talitlushäired.
Ravi. Sümptomaatiline.
24.2.8. Olivopontotserebellaarsed degeneratsioonid
Rühm närvisüsteemi pärilikke haigusi, mida iseloomustavad degeneratiivsed muutused väikeaju neuronites, alumiste oliivide tuumades ja aju sillas, mõnel juhul kaudaalse rühma kraniaalnärvide tuumades, vähemal määral seljaaju eesmiste sarvede, basaalganglionide radade ja rakkude kahjustus. Haigused erinevad pärilikkuse tüübi ja kliiniliste sümptomite erineva kombinatsiooni poolest. Koenigsmarki ja Weineri klassifikatsiooni järgi on olivopontotserebellaarset degeneratsiooni 5 tüüpi.
I tüüp - Mendeli olivopontotserebellaarne degeneratsioon Pärineb autosoomselt dominantselt. Kursus kulgeb aeglaselt. See võib ilmneda vanuses 11 kuni 60 aastat. Kliiniline pilt koosneb väikeaju kahjustuse sümptomitest (ataksia, lihaste hüpotensioon, lauldud kõne düsartria elementidega, tahtlik värisemine), kaudaalsete kraniaalnärvide tuumad (düsartria, düsfaagia), subkortikaalsed ganglionid (hüperkinees); püramidaalsed ja okulomotoorsed sümptomid on vähem levinud.
II tüüp – Fickler-Winkleri olivopontotserebellaarne degeneratsioon.Päris autosoomselt retsessiivselt. See avaldub vanuses 20–80 aastat väikeajukahjustuse sümptomitega, peamiselt jäsemete ataksiaga. Tundlikkus ja kõõluste refleksid ei muutu. Pareesi ei täheldata.
III tüüp - olivopontotserebellaarne degeneratsioon koos võrkkesta degeneratsiooniga Pärineb autosomaalselt dominantselt. Tekib noores eas. Koos väikeaju ja ekstrapüramidaalsete sümptomitega määratakse võrkkesta ganglionrakkude pigmentaarsest degeneratsioonist tingitud nägemisteravuse progresseeruv langus.
IV tüüp – Schut-Haykmani olivopontotserebellaarne degeneratsioon.Päris autosomaalselt dominantselt. See ilmneb lapsepõlves ja noorukieas. Lisaks väikeaju sümptomitele tuvastatakse VII, IX, X ja XII paari kraniaalnärvide tuumade (näonärvi halvatus, bulbar sümptomid) ja seljaaju tagumiste nööride kahjustused (lihas-liigese tunne ja vibratsioonitundlikkuse häired). .
V tüüp - olivopontotserebellaarne degeneratsioon koos dementsuse, oftalmopleegia ja ekstrapüramidaalsete häiretega. Pärandi tüüp on autosoomne dominantne. Areneb keskeas. Seda iseloomustavad dementsus, progresseeruv oftalmopleegia, ekstrapüramidaalsed ja väikeaju sümptomid.
Olivopontotserebellaarseid degeneratsioone tuleks eristada Friedreichi ja Pierre Marie pärilikust ataksiast, hulgiskleroosi progresseeruvatest vormidest, väikeaju kasvajatest ja parkinsonismi juveniilsetest vormidest.
Ravi. Sümptomaatiline. Viia läbi mittespetsiifilise taastava ravi kursused, massaaž, füsioteraapia harjutused.