Какие бывают заболевания мышц. Симптомы локальной формы миозита. Каковы основные неврологические проявления синдрома Гийена-Барре
Дегенеративные заболевания с преимущественным поражением периферических нервов и мышц составляют значительную долю наследственной патологии человека. Диагностика нервно-мышечных заболеваний основана на молекулярно-генетических и электрофизиологических (ЭМГ) исследованиях.
Электронейромиография позволяет подтвердить диагноз и следить за динамикой заболевания. При нейрогенной мышечной патологии можно выявить признаки денервации: потенциалы фибрилляции, положительные острые волны, снижение амплитуды интерференционного потенциала, полифазные потенциалы. При первичной мышечной патологии ЭМГ-картина неспецифична и вариабельна; наиболее характерно снижение амплитуды потенциалов. Показатели скорости проведения импульса (СПИ) при аксонопатиях уменьшены незначительно или находятся на нижней границе нормы. При демиелинизирующих невропатиях СПИ значительно уменьшена. По изменению СПИ и амплитуды потенциалов действия (по чувствительным или смешанным нервам) можно диагностировать туннельные ней- ропатии, а также дифференцировать аксонопатии и миелинопатии. Увеличение латентного периода поздних ответов наблюдается при невропатиях и корешковом синдроме.
Значительную роль в диагностике играют морфологические, иммуногистохимические и электронно-микроскопические методы изучения биоптатов. Состояние мышечных волокон при световой биомикроскопии помогает дифференцировать первичную миогенную атрофию от вторичной денервационной (неврогенной или миелогенной) амиотрофии. Гистохимический анализ биоптатов необходим для обнаружения специфических метаболических дефектов в мышечной ткани. Электронная микроскопия открыла целый класс заболеваний, которые объединены понятием «структурная миопатия».
Лечение. Для многих заболеваний мышц, нервно-мышечных синапсов, периферических нервов и мотонейронов разработано этиологическое и патогенетическое лечение. В остальных случаях терапия направлена на замедление прогрессирования заболевания, продление периода ремиссии и повышение качества жизни больного. Лечение нервно-мышечных заболеваний требует совместных усилий неврологов и реабилитологов. Тактика лечения зависит от тяжести и скорости прогрессирования заболевания.
Рис. 6.1. Внешний вид ребенка 13 лет, получавшего длительную гормональную терапию. Кушингоид
Принципы длительной кортикостероидной терапии
Осложнения зависят от дозы и длительности лечения (рис. 6.1). Основные осложнения: синдром Кушинга, сахарный диабет, остеопороз, активизация туберкулеза, артериальная гипертония, психоз, подверженность инфекциям, язвенная болезнь.
При отмене кортикостероидов возможны 3 вида осложнений. 1. Осложнения, связанные с угнетением функции надпочечни-
ков. Оно развивается при дробном приеме преднизолона в дозе, превышающей 20-30 мг/сут в течение более одной недели. Полное восстановление занимает до одного года. При дозах, приближенных к физиологическим, функция надпочечников обычно остается сохранной, если длительность лечения не превышает 1 мес. После обычных доз кортикостероидов заместительной терапии не требуется.
2. Общие симптомы отмены (анорексия, тошнота, рвота, сонливость, головная боль, лихорадка, миалгия и артралгия, потеря массы) более вероятны после длительной терапии. Лечение симптоматическое, малыми дозами кортизона (10 мг/сут) в течение нескольких недель.
3. Обострение основного заболевания. Это одно из наиболее опасных осложнений при отмене кортикостероидов. Его риск уменьшается при постепенном снижении дозы. При нервно-мышечных заболеваниях чаще всего применяется преднизолон - препарат короткого действия для приема внутрь. Его можно назначать ежедневно (в несколько приемов или однократно утром) или через день (однократно утром). При коротком курсе (менее месяца) схема приема не имеет существенного значения. При длительном лечении дробный ежедневный прием способствует развитию синдрома Кушинга, угнетению функции надпочечников и снижает сопротивляемость инфекциям. При длительном курсе однократный утренний прием суточной дозы препарата короткого действия реже вызывает угнетение над-
почечников (хотя и не предотвращает возникновение синдрома Кушинга). При приеме через день удвоенной суточной дозы реже развиваются угнетение надпочечников, синдром Кушинга и снижение сопротивляемости инфекциям. Эта схема эффективна при большинстве нервно-мышечных заболеваний.
6.1. Прогрессирующие мышечные дистрофии
Термином «мышечные дистрофии» обозначают группу клинически полиморфных генетически детерминированных заболеваний, в основе которых лежат первичные прогрессирующие дегенеративные изменения в мышечных волокнах. Различные формы миодистрофий отличаются друг от друга своей генетической природой, типом наследования, сроком дебюта, топографическим своеобразием распределения мышечных атрофий. К характерному клиническому маркёру миодистрофий относится «утиная» походка, связанная со слабостью ягодичных мышц, которые фиксируют таз относительно бедренной кости. В результате во время ходьбы возникают наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). Кроме того, у больных можно наблюдать ходьбу на пальцах, частые падения, медленное двигательное развитие и специфические ограничения при поднимании рук вверх, подъеме по лестнице, вставании с пола.
Миодистрофии Дюшенна и Беккера. Форма Дюшенна широко распространена в мире и встречается с частотой 1 на 3500 новорожденных мальчиков, тогда как форма Беккера наблюдается примерно в 3-5 раз реже.
Этиология и патогенез. Миодистрофии Дюшенна и Беккера являются аллельными вариантами, наследуются по рецессивному сцепленному с Х-хромосомой типу и обусловлены либо полным отсутствием синтеза, либо синтезом дефектного высокомолекулярного цитоскелетного белка-дистрофина. Из-за отсутствия дистрофина миофибриллы утрачивают устойчивость к циклическим актам сокращения-расслабления и разрываются. Саркоплазматические мембраны становятся нестабильными, нарушается работа ионных каналов, в результате повышается концентрация свободного внутриклеточного ионизированного кальция, который оказывает некротизирующее влияние на мышечные волокна, вызывая их лизис (рис. 6.2).
Клиническая картина. Первые клинические симптомы у большинства мальчиков с миодистрофией Дюшенна возникают до 3-5 лет жизни: нарушается походка, дети начинают часто падать, утрачивают

Рис. 6.2. Молекулярная организация дистрофина

Рис. 6.3. Больные, изображенные Г. Дюшенном
подвижность. Развивающаяся псевдогипертрофия икроножных мышц создает обманчивое впечатление о мышечной силе (рис. 6.3). Псевдогипертрофии могут развиваться также в ягодичных, дельтовидных мышцах, мышцах живота и языка. Наконец, мышечная слабость становится настолько явной, что ребенок с трудом встает с пола, ходит «утиной» походкой, использует миопатические приемы: «взбирание по себе», «подъем лесенкой» (симптомы Говерса).
Рис. 6.4. Ребенок 1,5 года с болезнью Дюшенна
Рис. 6.5. Тот же ребенок в 5 лет. Псевдогипертрофии мышц, лордоз
Двигательные функции относительно стабилизируются между 3 и 6 годами жизни. В большинстве случаев возможность ходить и подниматься по лестнице сохраняется до 8-летнего возраста. От 3 до 8 лет происходит дальнейшее укорочение ахилловых сухожилий и формируются фиксированные сгибательные контрактуры в голеностопных суставах, развиваются компенсаторный поясничный гиперлордоз, кифосколиоз грудного отдела позвоночника, нарастают атрофии мышц бедра, тазового, а затем и плечевого пояса, спины и проксимальных отделов рук. Обращает на себя внимание наличие «свободных надплечий», «крыловидных лопаток», «осиной талии». Нередко атрофии мышц маскируются хорошо развитым подкожным жировым слоем. Часто развиваются деформации грудной клетки и стоп, диффузный остеопороз. Коленные, сгибательные и разгибательные локтевые рефлексы исчезают в первую очередь, тогда как ахилловы рефлексы могут сохраняться довольно долго. В возрасте 9 лет некоторые дети уже передвигаются с помощью кресла-каталки, но у большинства способность к самостоятельному передвижению сохраняется вплоть до 12-летнего, а возможность стоять - до 16-летнего возраста. Слабость дыхательной мускулатуры и диафрагмы обусловливает уменьшение жизненной емкости легких до 20 % нормы, что приводит к эпизодам ночной гиповентиляции (рис. 6.4-6.6).
У части больных обнаруживаются различные признаки эндокринопатии: адипозогенитальный синдром, низкорослость. В связи

Рис. 6.6. Тот же ребенок в 14 лет. Выражены деформация позвоноч- ника, контрактуры сгибательного характера, атрофии мышц

Рис. 6.7. Псевдогипертрофии мышц голеней при болезни Беккера
с дефицитом церебральных изоформ дистрофина - аподистрофинов у части больных с миодистрофией Дюшенна имеет место умственная отсталость различной степени. Тяжесть психических нарушений у детей не соотносится с выраженностью мышечного дефекта и стадией миодистрофического процесса. Облигатным признаком развернутой стадии миодистрофии Дюшенна является гипертрофическая, или дилатационная, кардиомиопатия, которая сопровожда- ется нарушениями ритма сердца, расширением его границ, явлениями сердечной недостаточности. Кардиомиопатия - наиболее частая причина летального исхода при миодистрофии Дюшенна. К летальности приводит также дыхательная недостаточность, которая провоцируется интеркуррентными инфекциями или аспирацией. Больные погибают на 2-3-м десятилетии жизни.
Миодистрофия Беккера (рис. 6.7) может развиваться после 15-
20 лет, протекает гораздо мягче. Больные с этой формой миодистрофии доживают до зрелого возраста. Нарушения интеллекта для нее нехарактерны, ретракции сухожилий и контрактуры менее выражены, чем при миодистрофии Дюшенна, кардиомиопатия может отсутствовать. Однако у некоторых больных нарушения деятельности сердца выступают на первый план и часто являются манифестным симптомом болезни. Кроме того, у части больных миодистрофией Беккера сохранена фертильность, поэтому взрослые больные через дочь могут передавать заболевание своим внукам («эффект деда»).
Диагностика. Для миодистрофии Дюшенна характерно значительное повышение уровня ферментов уже на ранних стадиях миодистро-
фического процесса. У больных до 5-летнего возраста уровень креатинфосфокиназы (КФК) может превышать верхнюю границу нормы в десятки и даже сотни раз. Затем концентрация фермента снижается приблизительно на 20% в год. Содержание в сыворотке альдолазы, лактат-дегидрогеназы, трансаминаз также повышено. Высокая активность КФК - практически облигатный признак болезни и, кроме миодистрофии Дюшенна, может встречаться при миодистрофии Беккера (как правило, не превышая 5000 ЕД/л), полимиозите, дерматомиозите, гипотиреозе, алкогольной миопатии и пароксизмальной миоглобинурии. ЭМГ выявляет признаки первично-мышечного поражения (низковольтажная кривая с обилием полифазных потенциалов, укорочение потенциалов действия двигательных единиц).
В настоящее время общепринятым «золотым стандартом» для диагностики миодистрофии Дюшенна и Беккера, выявления носи- тельства гена и пренатальной диагностики является мутационный анализ. Иммуногистохимическая реакция на дистрофин применяется при анализе процентного содержания дистрофина в мышцах и отличает формы Дюшенна и Беккера (при первой он отсутствует). У гетерозиготных носительниц (матерей и сестер больных) примерно в 70% случаев выявляются субклинические признаки патологии скелетных мышц: повышение КФК, первично-мышечные изменения на ЭМГ и при исследовании мышечных биоптатов. Изредка у носительниц отмечаются уплотнение и увеличение в объеме икроножных мышц, повышенная мышечная утомляемость при физической нагрузке, спазмы в мышцах после нагрузки (крампи).
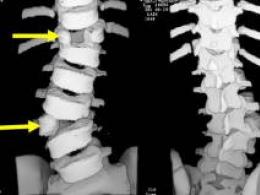
Рентгенография костей помогает выявить атрофию диафизов длинных трубчатых костей, истончение кортикального слоя, сужение костно-суставного канала, диффузный остеопороз.
Поражение сердечно-сосудистой системы (кардиомиопатия) развивается у 73% больных детей. Недостаточность дистрофина в кар- диомиоцитах приводит к прогрессирующей атрофии кардиомиоцитов и замещению их фиброзной тканью. Кардиомиопатия впервые диагностируется в 6-7 лет, к 20 годам она имеется у 95% больных. Отмечаются также тахикардия, аритмия, лабильность пульса и АД, приглушение тонов, расширение границ сердца. На ЭКГ отмечаются нарушения сердечного ритма, желудочковые экстрасистолы, признаки гипертрофии левого желудочка (27%): глубокий зубец Q в отведениях II-III aVF и V 6 ; высокий R в отведении V 1 , признаки ишемии миокарда (5%). На Эхо-КГ можно выявить гипертрофическую (55%) или дилатационную
(25%) кардиомиопатию, дефект межпредсердной перегородки, пролапс митрального клапана, миксому левого желудочка.
Биопсия сердечной мышцы обнаруживает атрофию мышечных волокон, интерстициальный фиброз, жировую инфильтрацию.
Дифференциальная диагностика миодистрофии Дюшенна и Беккера проводится с врожденной дисплазией тазобедренных суставов, витамин- D-резистентным рахитом, проксимальными типами спинальных амиотрофий, полимиозитом и дерматомиозитом, метаболическими и эндокринными миопатиями.
При наличии клинического фенотипа миодистрофии Дюшенна у девочек следует в первую очередь исключить наличие Х-аутосомных транслокаций или других хромосомных аберраций с заинтересован- ностью Х-хромосомы, а также некоторые другие редкие генетические варианты. Кроме того, требуется исключить синдром Шерешевского- Тернера (Х-моносомия). С этой целью проводят цитогенетическое исследование кариотипа.
Миодистрофия Эмери-Дрейфуса является медленно прогрессирующей формой миодистрофии с X-сцепленным рецессивным типом наследования, которая обусловлена мутацией в гене цитоскелетного мышечного белка - эмерина, продуцирующегося преимущественно в скелетных, гладких мышцах и кардиомиоцитах.
Клиническая картина (рис. 6.8). Заболевание начинается между 5 и 15 годами жизни. Самыми ранними и типичными симптомами являются нарастающие сгибательные контрактуры в локтевых суставах и разгибателях кистей, ретракции ахилловых сухожилий. Как правило, в 12-летнем возрасте у пациентов уже значительно выражены контрактуры в коленных, голеностопных и локтевых суставах. Затем возникают слабость и атрофия двуглавых и трехглавых мышц плеча, позже - дельтовидных и других мышц плечевого пояса. В некоторых случаях в качестве первого симптома отмечают ходьбу на пальцах и наружных краях стоп, которая возникает приблизительно в 5-летнем возрасте. До этого момента двигательное развитие детей обычно не нарушено. Мышечная слабость возникает незаметно и медленно прогрессирует. Примерно в 20-летнем возрасте наступает относительная стабилизация. Способность к ходьбе и подъему по лестнице сохраняется. Лицевая мускулатура не страдает. Слабость мышц представлена в руках (лопаточно-плечевая) и в ногах (перонеальная). Приемы Говерса и псевдогипертрофия икроножных мышц могут отсутствовать. Сухожильные рефлексы не вызываются. Часто укорочены заднешейные мышцы, отмечается ограничение

Рис. 6.8. Пациентка 12 лет с миодистрофией Эмери-Дрейфуса
движений в шейном отделе позвоночника (синдром ригидного позвоночника). Частыми и прогностически важными симптомами болезни являются нарушения сердечной проводимости и развивающаяся дила- тационная или гипертрофическая кардиомиопатия. Кардиомиопатия может осложняться развитием паралича предсердий вследствие фиброза пейсмекеров синусового узла. В этих случаях показана срочная имплантация искусственного водителя ритма.
Синкопальные состояния и приступы брадикардии в некоторых случаях могут предшествовать появлению мышечной слабости, но чаще возникают на 3-м десятилетии жизни. Изменения в проводящей системе сердца далеко не всегда обнаруживаются при стандартном ЭКГ-исследовании, но при мониторировании можно выявить атриовентрикулярные блокады и периоды Самойлова-Венкебаха. Аритмия, которую не удается скорригировать имплантацией искусственного водителя ритма, может привести к инсульту и смерти больного. Витальный прогноз при миодистрофии Эмери-Дрейфуса всецело зависит от степени поражения сердца.
Диагностика. Активность КФК повышена умеренно, лактатде- гидрогеназы и альдолазы - в меньшей степени. В пользу миодистрофии Эмери-Дрейфуса свидетельствует отсутствие иммунофлюоресцентной реакции на эмерин с 12 моноклональными антителами при биомикроскопии лейкоцитов, мышечных и кожных биоптатов. Для болезни характерны сочетанные ЭМГ-признаки первично-мышечного и неврогенного поражения с большой представленностью спонтанной денервационной активности.
Лице-лопаточно-плечевая миодистрофия (тип Ландузи-Дежерина). Заболевание наследуется по аутосомно-доминантному типу с высокой пенетрантностью и вариабельной экспрессивностью. Встречается с частотой 2,9 на 100 000 населения. Установлена генетическая гетерогенность лице-лопаточно-плечевой миодистрофии. Большинство случаев связано с мутацией длинного плеча хромосомы 4.
Клиническая картина. Заболевание обычно начинается на 2-м десятилетии жизни. Первоначально атрофии наблюдаются в плечевом поясе, позже распространяются на лицо. У больных обедняется мимика; речь становится неразборчивой. На высоте заболевания поражаются круговые мышцы рта и глаз, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая и трехглавая мышцы плеча. Отмечаются характерные симптомы в виде «поперечной улыбки» («улыбки Джоконды»), протрузии верхней губы («губы тапира»). Грудная клетка уплощается в переднезаднем направлении, плечевые суставы ротированы внутрь, лопатки приобретают крыловидную форму. Атрофии распространяются в нисходящем направлении. При вовлечении в процесс мышц ног слабость наиболее заметна в перонеальной группе мышц - «свисающая стопа». Характерна асимметрия атрофии. Может наблюдаться псевдогипертрофия мышц. Контрактуры и ретракции сухожилий выражены умеренно. Кардиомиопатия редка. Аномалии сосудов сетчатки при ангиоретинографии рассматривают как одно из фенотипических проявлений болезни. Тяжелые глазные симптомы сопровождаются теле- ангиэктазией, отеком и отслойкой сетчатки. Может наблюдаться снижение слуха. Телеангиэктазии ликвидируют коагуляцией, что предотвращает развитие слепоты. Течение болезни относительно благоприятно. Физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни. Многие больные
сохраняют работоспособность, и качество их жизни не ухудшается. Других больных болезнь приковывает к креслу-каталке в зрелом возрасте.
Диагностика. Уровень КФК может повышаться в 5 раз. При ЭМГ регистрируются как миопатические двигательные единицы, так и денервационные потенциалы. Во многих мышцах конечностей гистологические изменения минимальны; в надлопаточных мышцах обнаруживают прогрессирующую дегенерацию и краевую денервацию. Необходимо исключить миастению и опухоль мозгового ствола.
Конечностно-поясные миодистрофии (КПМД) - случаи проксимальной мышечной слабости, которая начинает развиваться на 2-м или 3-м десятилетии жизни, медленно прогрессирует и приводит к глубокой инвалидизации лишь через 15-20 лет.
Этиология и патогенез. КПМД не является однородной в генетическом отношении; на сегодняшний день обнаружено около 10 различных генетических дефектов.
Клиническая картина. Первыми поражаются мышцы плечевого и тазового пояса. В развернутых стадиях значительно поражаются мышцы спины и живота, формируется поясничный гиперлордоз. Мышцы лица, как правило, не страдают. У больных обнаруживаются типичная «утиная» походка, миопатические приемы. Контрактуры и псевдогипертрофии мышц нехарактерны. Кардиомиопатии не развиваются; интеллект сохранен. Мужчины и женщины поражаются одинаково часто. Летальный исход может наступать от легочных осложнений.
Диагностика. Содержание КФК умеренно повышено. ЭМГ демонстрирует признаки первично-мышечного поражения. КПМД необходимо отличать от миопатии Беккера, ювенильной спинальной амиотрофии, миопатии с накоплением гликогена, эндокринных, токсических, лекарственных миопатий, полимиозита и миозита.
6.2. Врожденные структурные миопатии
Врожденные структурные миопатии (ВСМ) представляют собой генетически гетерогенную группу медленно прогрессирующих заболеваний скелетных мышц. Клиническая симптоматика различных ВСМ неспецифична. Основной клинический симптом - диффузная мышечная гипотония, которая может возникать еще внутриутробно и определять редкое шевеление плода. ВСМ принадлежит значительный удельный вес среди причин так называемого синдрома вялого ребенка. Гипотония превалирует в мышцах тазового пояса и прок-
симальных отделов ног. Мышцы плечевого пояса и рук поражаются в меньшей степени. Нередко выявляются врожденный вывих бедра, долихоцефалическая форма головы, готическое нёбо, конская стопа, кифосколиоз, гипоплазия мышц. Характерна задержка двигательного развития: дети поздно начинают держать голову, сидеть, вставать, ходить, часто падают при ходьбе, неспособны бегать. В дальнейшем они не могут выполнить простейших гимнастических упражнений, участвовать в подвижных играх. Сухожильные рефлексы у больных могут быть нормальными, сниженными или отсутствовать. Чрезвычайно важным критерием ВСМ является отсутствие прогрессирования или очень медленное нарастание мышечной слабости. При некоторых формах двигательные функции с возрастом могут несколько улучшаться.
Диагностика. Активность КФК в норме или чуть повышена. При ЭМГ регистрируются низкоамплитудные полифазные миопатические потенциалы двигательных единиц. Скорость проведения импульса по двигательным и чувствительным волокнам в норме. Диагноз достоверно устанавливается лишь при проведении мышечной биопсии с использованием световой и электронной микроскопии, кото- рая обнаруживает специфическое строение мышечного волокна. Исследование мышечных биоптатов у больных детей может выявить уникальные гистологические признаки, определившие целый ряд наименований: болезнь центрального стержня, миотубулярная миопатия, немалиновая миопатия, трехпластинчатая миопатия, миопатия с лизисом волокон I типа, миопатия со сферическими тельцами, миопатия с накоплением телец в виде «отпечатков пальцев», миопатия с цитоплазматическими включениями в виде редуцированных телец, миопатия с агрегацией трубочек и т.д.
Лечение мышечных дистрофий. Терапевтические возможности при миодистрофиях значительно ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено на как можно более длительное поддержание имеющейся мышечной силы, снижение темпа развития атрофии и предотвращение формирования контрактур. Основная задача состоит в том, чтобы на максимально возможный срок продлить период активности.
Комплексное лечение состоит из медикаментозной терапии, физиотерапевтических процедур, лечебной гимнастики и массажа, ортопедической коррекции и соблюдения диеты. Важную роль играют психологическая поддержка, продолжение обучения, правильная профессиональная ориентация.
Физиотерапевтические процедуры включают электрофорез прозерина, хлорида кальция, синусоидально-модулированные или диадинамические токи различной проникающей способности, электромиостимуляцию, озокеритовые, парафиновые и грязевые аппликации, ванны (радоновые, хвойные, серные, сероводородные). Рекомендуется оксибаротерапия, поскольку кислород ингибирует процессы фиброзирования и коллагенообразования. Ортопедическая коррекция консервативного (специальные шины и укладки) и оперативного характера (ахиллотомия, миотомия) направлена на борьбу с контрактурами и формирующимися патологическими установками конечностей и также имеет цель сохранить способность больного к самостоятельному передвижению. В каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. При развивающихся контрактурах после проведения тепловых процедур рекомендуется осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Больному рекомендуется диета, обогащенная белком, с ограничением жиров (особенно животного происхождения) и углеводов при оптимальном и сбалансированном содержании витаминов и микроэлементов. Необходимо избегать соленого, жареного, пряностей, маринадов, крепких мясных бульонов, кофе, шоколада, какао, пирожных, сдобного теста.
Медикаментозная терапия преследует цель компенсировать энергетический дефицит в мышечной ткани, улучшить тканевый метаболизм и кровообращение, стабилизировать мембраны мышечных волокон. Применяют никотиновую кислоту, витамины В 6 , В 12 , А и Е (аевит). Для улучшения белково-синтетических процессов используют аминокис- лотные препараты (церебролизин, глицин, метионин, глутаминовую, фолиевую кислоты). Назначают нестероидные анаболические средства (калия оротат), макроэргические препараты (фосфаден), кардиотрофики (рибоксин, карнитина хлорид, солкосерил), средства, улучшающие периферическое кровообращение (трентал, галидор, теоникол, оксибрал) и ноотропы [пантогам, пирацетам (ноотропил)]. Для улучшения энергетических процессов, происходящих в системе дыхательной цепи митохондрий, используют коэнзим Q10 (убихинон), лимантар, внутривенные инфузии цитохрома-С. Эффекты дезинтоксикации и улучшения реологических свойств крови, купирование слайдж-синдрома достигаются инфузиями вазоактивных препаратов, реополиглюкина, курсов плазмафереза. Относительной стабилизации клеточных мембран способствуют малые дозы преднизолона. Для коррекции
кардиомиопатии используют кардиотрофики (кроме больных с гипертрофической кардиомиопатией); при сердечной недостаточности - сердечные гликозиды, диуретики, каптоприл. При сердечных аритмиях назначают хинидин, β-адреноблокаторы, антагонисты кальция. При развитии полной атриовентрикулярной блокады актуальным становится вопрос о целесообразности имплантации искусственного электрокардиостимулятора.
Перспективы разработки методов генетической терапии при некоторых миодистрофиях (болезнях Дюшенна, Беккера) связаны с совершенствованием генетических технологий. Идет активный поиск генети- ческих носителей (векторов), способных встроить ген дистрофина или мини-гены в мышечные клетки больного реципиента. Исключительное значение придается медико-генетическому консультированию семьи, проведению пренатальной диагностики с исследованием ДНК плода.
6.3. Спинальные мышечные амиотрофии
Спинальные мышечные амиотрофии (СМА) - это гетерогенная группа наследственных заболеваний периферической нервной системы. Патогенез связан с прогрессирующей дегенерацией мотонейронов передних рогов спинного мозга (в некоторых случаях и двигательных ядер ствола мозга). Причиной этого является генетический дефект, вызывающий программируемую клеточную гибель - апоптоз клеток. Утрата мотонейронов приводит к развитию вялого паралича и денервационной атрофии поперечнополосатых мышц. В большинстве случаев наблюдается симметричное поражение проксимальных мышц конечностей; дистальные амиотрофии, поражение буль-
барной мускулатуры и асимметрия поражения развиваются реже. Центральный мотонейрон, как правило, интактен. Расстройств чувствительности не бывает.
Разные варианты СМА отличаются возрастом дебюта, характером течения, топографией поражения скелетной мускулатуры и типом наследования (рис. 6.9). Большинство форм наследуется по аутосомно-рецессивному типу. Несколько форм характеризуются

Рис. 6.9. Синдром вялого ребенка при СМА
аутосомно-доминантным и Х-сцепленным рецессивным типами наследования. При гистологическом исследовании мышечных биоптатов выявляют, что к группам волокон обычного размера прилежат мышечные волокна малого размера, пучки гипертрофированных и атрофичных мышечных волокон.
Если ЭМГ определяет бесспорные симптомы СМА, проведение мышечной биопсии необязательно. Принципы лечения и реабилитации СМА такие же, как при миодистрофиях. Этиотропное и патогенетическое лечение пока не разработано.
Проксимальные спинальные амиотрофии детского возраста наследуются по аутосомно-рецессивному типу. Выделяют три фенотипи- чески различных варианта, отличающихся возрастом клинической манифестации, течением и прогнозом:
Тип I, или острая злокачественная инфантильная СМА Верднига- Гоффмана;
Тип II, или хроническая инфантильная СМА (промежуточный тип);
Тип III, или ювенильная СМА Кугельберга-Веландера.
В их основе лежит единая генетическая мутация - делеция гена жизнеспособности моторного нейрона, расположенного на длинном плече хромосомы 5. Поиск мутации проводят при ДНК-диагностике, в том числе у плода при пренатальной диагностике, которая помогает избежать рождения больного ребенка.
Острая злокачественная инфантильная спинальная амиотрофия (болезнь Верднига-Гоффмана, или СМА I типа) встречается с частотой 1 на 25 500 новорожденных. Клинические симптомы отмечаются уже при рождении или появляются до 6 мес жизни. Еще внутриутробно отмечается вялое шевеление, свидетельствующее о снижении двигательной активности плода. У больного ребенка обнаруживают генерализованную слабость, преимущественно в проксимальных мышечных группах, гипотонию и арефлексию. В положении на спине наблюдается «поза лягушки» с разведением и наружной ротацией бедер. Мимическая мускулатура относительно сохранна, глазодвигательные мышцы не вовлечены. Дыхательная функция поначалу адекватная. Выявляются атрофия и фасцикуляции в языке, фасцикулярный тремор кистей. При развитии бульбарного синдрома исчезает глоточный рефлекс, значительно затрудняется кормление, что может привести к аспирационной пневмонии. Часто формируется деформация грудной клетки (рис. 6.10). Если мышечная слабость

Рис. 6.10. Ребенок, 6 мес, с болезнью Верднига-Гоффмана
выявляется сразу после рождения, то смерть наступает приблизительно в 6-месячном возрасте, тогда как при появлении первых симптомов после 3 мес жизни срок выживаемости может составлять около 2 лет. Основная причина смерти - дыхательная недостаточность на фоне интер- куррентных респираторных заболеваний (рис. 6.11, 6.12).
Для диагностики выявляют мутацию гена путем молекулярно-генетического анализа. Концентрация КФК обычно в норме, но у детей с быстро прогрессирующей слабостью она может быть несколько повышена. ЭМГ обнаруживает потенциалы фибрилляций и фасцикуляции в покое и повышение средней амплитуды потенциалов двигательных единиц. Скорость проведения по двигательным аксонам периферических нервов, как правило, соответствует норме. СМА I типа необходимо дифференцировать от других состояний, вызывающих синдром «вялого ребенка». К ним относятся врожденные миодистрофии и невропатии, структурные миопатии, врожденная или неонатальная миастения, метаболические миопатии, внутриутробный полиомиелит, ботулизм, хромосомная патология, атоническая форма церебрального паралича, синдром Марфана.
Хроническая инфантильная спинальная амиотрофия (СМА II типа). Мышечная слабость, как правило, появляется между 6-м и 24-м мес жизни. Чем раньше дебютируют симптомы, тем злокачественнее течение. Первоначальные проявления слабости обычно симметричны и наблюдаются в проксимальных мышечных группах конечностей. Слабость мышц бедер - наиболее заметный симптом. В раннем периоде дистальная мышечная слабость минимальна или отсутствует. Сухожильные рефлексы с пораженных мышц резко снижены. Все больные способны сидеть, большинство - самостоятельно стоять, а некоторые даже могут ходить (рис. 6.13). Мимическая мускулатура

Рис. 6.11. Мальчик, 5 лет, с болезнью Верднига-Гоффмана

Рис. 6.12. Мальчик, 3 лет, с болезнью Верднига-Гоффмана

Рис. 6.13. Девочка, 9 лет, с болезнью Кугельберга-Веландера
и наружные мышцы глаза на ранних этапах болезни не поражаются. Мышечная слабость прогрессирует медленно. В отдельных случаях она остается стабильной многие годы, а затем прогрессирование воз- обновляется. Предполагается выживание больных вплоть до зрелого возраста, но даже в период относительной стабилизации ЭМГ выявляет
потенциалы фибрилляций и фасцикуляций. Формируются контрактуры, эквиноварусная деформация стоп. Уже в грудном возрасте у детей наблюдаются искривление позвоночника, деформации грудной клет- ки, дисплазия тазобедренных суставов.
Диагностика. Концентрация КФК в норме. Результаты генетического анализа и данные ЭМГ те же, что и при острой инфантильной форме.
Ювенильная спинальная амиотрофия (болезнь Кугельберга-Веландера, или СМА III типа) встречается в общей популяции с частотой 1,2 на 100 000. Двигательная активность во внутриутробном периоде достаточна; при рождении ребенок здоров. Дебют симптомов происходит между 2-м и 15-м годом жизни. Дети начинают неустойчиво ходить из-за нарастающей проксимальной мышечной слабости в ногах. Развиваются псевдогипертрофии икроножных мышц, что часто ведет к ошибочной диагностике миодистрофии Дюшенна. Заболевание течет доброкачественно, прогрессирует очень медленно. Кисти поражаются позже. Лицевые мышцы могут быть ослаблены, но движения глазных яблок всегда в полном объеме. Бульбарные нарушения нехарактерны. Примерно у половины больных могут развиваться костные деформации, изредка - сухожильные ретракции и контрактуры в суставах. Сухожильные рефлексы с ослабленных мышц отсутствуют или значительно угнетены. Часто регистрируется фасцикулярный тремор кистей.
Диагностика. Первостепенное значение имеет выявление генетической мутации. Концентрация КФК может превышать верхнюю границу нормы в 2-4 раза. У половины больных при ЭМГ регистрируется спонтанная активность (фасцикуляции, фибрилляции и положительные острые волны). При напряжении мышц отмечаются повышение амплитуды и полифазия, увеличение длительности и снижение числа потенциалов двигательных единиц. Проведение по чувствительным волокнам нервов всегда в норме. Скорость проведения по двигательным волокнам при длительном течении заболевания может уменьшаться. СМА III типа дифференцируют с конечностнопоясными миодистрофиями.
Бульбоспинальная амиотрофия Кеннеди - редкая Х-сцепленная рецессивная форма СМА, дебютирующая на 4-й декаде жизни; изредка отмечаются случаи дебюта симптомов в 12-15 лет. Ген картирован на длинном плече Х-хромосомы. Мутация затрагивает ген андрогенового рецептора, в том числе спинальных мотонейронов, что делает эти
рецепторы нечувствительными к влияниям мужских половых гормонов (андрогенов). Ядро клинической картины составляют слабость, атрофии и фасцикуляции в проксимальных мышечных группах конечностей, сухожильная арефлексия, лицевая слабость, атрофии и фасцикуляции в языке, периоральные фасцикуляции, дизартрия и дисфагия, тремор и болезненные мышечные спазмы (крампи). Изредка развивается аксональная невропатия. Бульбарные наруше- ния возникают, как правило, спустя 10 лет после начала болезни. Характерны эндокринные нарушения: гинекомастия, тестикулярная атрофия, снижение потенции и либидо, сахарный диабет, бесплодие, обусловленное азооспермией. Прогноз болезни в целом благоприятен: сохраняются способность к ходьбе и возможность самообслуживания. Продолжительность жизни не уменьшается, однако повышен риск возникновения злокачественных новообразований вследствие гормонального дисбаланса (в том числе рака грудной железы).
Диагностика. В настоящее время возможно проведение прямой ДНК-диагностики, установление гетерозиготного носительства и осуществление дородовой диагностики. ЭМГ выявляет признаки денервации. Уровень КФК может быть в норме. Заболевание необходимо отличать от бокового амиотрофического склероза.
6.4. Множественный врожденный артрогрипоз
Множественный врожденный артрогрипоз - синдром, основным проявлением которого является ограничение подвижности в суставах в сочетании с их деформациями. Обычно поражаются дистальные суставы (голеностопные, лучезапястные), реже - коленные и локтевые суставы. Мышечная слабость при артрогрипозе может носить как неврогенный, так и миогенный характер. Подавляющее большинство случаев относится к спорадическим, остальные случаи наследуются по аутосомно-рецессивному или X-сцепленному типу. При неврогенном артрогрипозе наиболее активная фаза болезни наблюдается во внутриутробном периоде и уже в периоде новорожденности нарушены дыхание и глотание; часть детей погибает от аспирации. В более легких случаях выживаемость выше, а мышечная слабость прогрессирует очень медленно или не прогрессирует вообще. Дыхательные расстройства и проблемы с кормлением в дальнейшем исчезают. Контрактуры представлены как в проксимальных, так и в дистальных суставах. У некоторых новорожденных определяются сопутствующие микрогнатия, высокое нёбо, лицевые аномалии, как при
синдроме Эдвардса (трисомии 18). У некоторых детей с неврогенным артрогрипозом обнаруживаются аномалии развития переднего мозга. Бывают сочетания с менингомиелоцеле, микроцефалией и задержкой психического развития. Синдром миогенного артрогрипоза может наблюдаться при миопатии с диспропорцией типов волокон, врожденных миодистрофиях, миотонической дистрофии, миастенических синдромах, дефиците фосфофруктокиназы.
Диагностика. Гистологическое исследование мышц выявляет характерные признаки денервации и реиннервации. Также выявляют проявления миопатии: повышение доли коллагеновых волокон и жировой ткани, хаотичность расположения волокон среднего размера, фиброз капсул мышечных веретен.
6.5. Воспалительные миопатии
Дерматомиозит является системной иммунозависимой ангиопатией, при которой наблюдаются сосудистые окклюзии и инфаркты, приводящие к развитию всех характерных патологических изменений в мышцах, соединительной ткани, коже, желудочно-кишечном тракте и нервных волокнах. Патогенез связан с образованием антител и иммунных комплексов и активацией системы комплемента. В состав периваскулярного инфильтрата входят Т-лимфоциты, которые в подавляющей своей части являются Т-хелперами, В-лимфоциты и плазматические клетки.
Клиническая картина. Пик заболеваемости приходится на возраст 5-10 лет, но описаны случаи и более раннего начала (до 4-месячно- го возраста). Симптомы возникают постепенно или молниеносно. Скрытое начало характеризуется лихорадкой, недомоганием и утратой аппетита (анорексией). Мышечная слабость в это время может отсутствовать. Такие неспецифические симптомы сохраняются в течение недель и месяцев, что наводит на мысль о персистирующей инфекции. У большинства детей дерматит появляется раньше миозита. Сыпь вначале локализуется на верхних веках и имеет вид
эритемы с очагами нарушенной пигментации и отеком. Затем она распространяется вокруг глаз и на область щек. Эритема и отек на разгибательных поверхностях межфаланговых, локтевых и коленных суставов развиваются позже. Со временем кожа становится атрофичной и шелушащейся. Миопатические изменения включают проксимальную слабость, мышечную ригидность и боль. Слабость нарастает, быстро развиваются сгибательные контрактуры и деформации суставов. Сухожильные рефлексы снижаются, а затем исчезают. У 60% больных обнаруживаются кальцификаты в подкожной клетчатке, особенно под теми областями кожи, где нарушена пигментация. Множественные кальцификаты создают эффект «брони» при рентгенографии. У некоторых детей ведущим начальным симптомом является мышечная ригидность, а кожные и миопатические симптомы выражены не столь ярко. Инфаркты желудочно-кишечного тракта в терминальных стадиях болезни в прошлом приводили к смерти. Летальность при дерматомиозите в настоящее время снизилась и составляет менее 5%, что связано с совершенствованием методов лечения. Более чем у 30% взрослых с дерматомиозитом в дальнейшем выявляются злокачественные новообразования.
Диагностика. Сочетание лихорадки, сыпи, миалгии и слабости свидетельствует в пользу диагноза дерматомиозита. В начале болезни уровень КФК обычно повышен. Во время активного дерматомиозита ЭМГ покоя выявляет фибрилляции и положительные острые волны; при мышечном напряжении регистрируются укороченные низкоамплитудные полифазные потенциалы. Мышечная биопсия обнаруживает атрофию миофибрилл. Капиллярные некрозы вначале возникают по периферии мышечного пучка и вызывают ишемию прилежащих миофибрилл. Наиболее выражена атрофия в пучках, которые соприкасаются с большими фасциальными футлярами. Волокна I и II типов (тонические и фазические) поражаются в равной степени.
Лечение. Воспалительный процесс активен в течение 2 лет. Кортикостероиды снижают его активность, способствуя уменьшению симптоматики. Наилучшие результаты достигаются, когда кортикостероиды назначают на ранних стадиях болезни, в высоких дозах и применяют длительно. Препаратом выбора служит преднизолон. Начальная его доза дается из расчета 2 мг/кг в сутки, но не выше 100 мг/сут. Температура тела часто нормализуется в течение первых 48 ч от момента начала терапии. Иногда уровень КФК возвращается
к норме на 2-й нед лечения параллельно с заметным увеличением силы мышечного сокращения. В этом случае дальнейший прием преднизолона может осуществляться по схеме через день и в той дозе, которая позволит снизить тяжесть побочных эффектов стероидной терапии. Терапия преднизолоном одинаково эффективна при приеме препарата ежедневно или по схеме через день, но только в тех случаях, когда лечение не прерывается. Когда сила мышц нарастает, начальную дозу принимаемого через день преднизолона можно снижать на 10% в месяц в течение 5 мес. Дальнейшее уменьшение дозы преднизолона допустимо лишь на 5% в месяц. При решении вопроса о снижении дозы кортикостероидов недопустимо ориентироваться лишь на снижение активности КФК, поскольку заметное увеличение мышечной силы наступает только через 1-2 мес после уменьшения уровня фермента, т.е. ведущим критерием снижения дозы кортикостероидов служит позитивная клиническая динамика. У большинства больных поддерживающая доза преднизолона, принимаемого по схеме через день, которая необходима для нормализации силы мышечного сокращения и концентрации КФК, составляет 25% стартовой дозы.
При лечении преднизолоном у некоторых больных сыпь полностью исчезает, но у большинства остаются рубцовые изменения кожи. Длительная стероидная терапия требует контроля функции желудочно-кишечного тракта. Для защиты слизистой оболочки желудка назначают препараты хлорида калия и блокаторов Н 2 -рецепторов. Серьезным осложнением длительной терапии является развитие стероидной миопатии, которая может быть расценена как обострение основного заболевания. Отличить развивающуюся стероидную миопатию от обострения дерматомиозита по клиническим критериям довольно сложно. При стероидной миопатии, как правило, страдают проксимальные отделы конечностей, развиваются выраженные атрофии, активность КФК не увеличивается. У большинства детей с дерматомиозитом при лечении уже через 3 мес отмечается улучшение, но терапию преднизолоном необходимо продолжать в течение 2 лет. Если лечение прервано преждевременно, неизбежны рецидивы, развиваются кальциноз и контрактуры. Медикаментозное лечение дополняют физической реабилитацией, необходима дыхательная гимнастика. Массаж в активной фазе противопоказан. При правильном лечении благоприятный исход наблюдается у 80% детей с дермато- миозитом. При резистентности или непереносимости преднизолона
показано пероральное назначение цитостатиков: метотрексат в дозе от 10 до 20 мг/м 2 поверхности тела 2 раза в неделю или азатиоприн в дозе 50-150 мг/сут. Во время терапии необходим регулярный контроль за функцией печени и клеточным составом крови. Комбинация кортикостероидов и цитостатиков позволяет избежать длительной терапии преднизолоном в высоких дозах. В случаях, когда прием кортикостероидов ограничен их побочным действием, применяют плазмаферез или курс внутривенных инфузий иммуноглобулина. В неактивной стадии обострений обычно не возникает.
Полимиозит. Этиология в большинстве случаев остается неиз- вестной. Предполагается, что в патогенезе играют роль клеточные и гуморальные механизмы, что подтверждается нередким развитием заболевания на фоне аутоиммунных процессов (системная красная волчанка, узелковый периартериит, ревматоидный артрит, склеродермия), а также хорошим эффектом применения кортикостероидов и иммуносупрессоров. Патогенез связан с клеточно-опосредованной цитотоксической реакцией, реализуемой Т-лимфоцитами, сенсибилизированными к поверхностным антигенам мышечных волокон.
Клиническая картина. Полимиозит обычно возникает в зрелом возрасте (45-55 лет), у детей и подростков встречается редко и не связан со злокачественными новообразованиями. Постепенно, исподволь нарастает симметричная проксимальная мышечная слабость, лихорадка и миалгии нетипичны. Часто развивается слабость сгибателей шеи («свисающая голова»). Для заболевания характерны дисфагия и приступы удушья. Постепенно слабость распространяется и на дистальные отделы конечностей. Выраженность парезов варьирует, а в тяжелых случаях развивается тетраплегия. Изредка слабость ограничивается дистальными группами мышц, мышцами глаза или лица. У больного могут наблюдаться периоды стабилизации и даже ремиссии, что может приводить к ошибочной диагностике конечностно- поясной миодистрофии. При хроническом течении болезни постепенно нарастают мышечные атрофии; возможно формирование контрактур. Сухожильные рефлексы вызываются на ранних стадиях болезни и снижаются при уменьшении мышечной массы, но полностью никогда не исчезают. Этот важнейший дифференциально- диагностический признак позволяет исключить полиневропатию. Иногда заболевание начинается остро с общего недомогания; резкая мышечная слабость развивается в течение нескольких дней, появляются боли в мышцах плечевого пояса. Атрофии мышц очень легкие
или отсутствуют. Часто при рентгенографии в мышцах обнаруживают кальцификаты. У взрослых типичны сердечно-легочные осложнения, нехарактерные для детской формы болезни.
Диагностика. Изменения КФК редки. ЭМГ-исследование практически всегда выявляет типичные признаки и миопатического, и неврогенного процессов. Мышечная биопсия обнаруживает различные патологические отклонения. Гистологически периваскулярная воспалительная инфильтрация наблюдается не всегда, поэтому отсутствие клеточных инфильтратов в образцах биоптатов не исключает диагноз полимиозита.
Для лечения полимиозита используется такая же схема, как при дерматомиозите. Больным, которые резистентны к кортикостерои- дам, показаны цитостатики (метотрексат). Плазмаферез и внутривенные введения иммуноглобулина являются оправданными альтернативными методами лечения при недостаточной эффективности общепринятой терапии.
Острый инфекционный миозит возникает после перенесенного гриппа или другой респираторно-вирусной инфекции. Симптомы вирусной инфекции сохраняются от 1 до 7 дней, а затем появляется интенсивная симметричная боль и слабость в мышцах. В тяжелых случаях за 1 сут больной становится обездвиженным. На фоне общей слабости проксимальные мышечные группы поражены тяжелее, чем дистальные. Болезненна пальпация мышц. Сухожильные рефлексы сохраняются. Уровень КФК обычно более чем в 10 раз превышает верхнюю границу нормы. Почти немедленно вслед за развитием миозита наблюдается его спонтанное обратное развитие. В худшем варианте для исчезновения болевого синдрома требуется от 2 до 7 дней постельного режима, после чего больной полностью выздоравливает.
Миотония. Феномен миотонии представляет собой замедленную реакцию расслабления (релаксации) мышцы после ее сокращения. Выделяют миотонию действия, перкуссионную или механическую миотонию и электромиографическую миотонию.
В патогенезе миотонии играет роль нестабильность мембраны мышечного волокна, что приводит к повторным сокращениям мышцы в ответ на одиночный раздражитель. Повторные миотонические импульсы возникают не спонтанно, а всегда при внешнем воздействии или в результате произвольного сокращения. Миотонию действия можно наблюдать у больного после интенсивного сокращения мышцы. Больного просят, например, сильно сжать кисть в

Рис. 6.14. Миотонические феномены у ребенка с миотонией Томсена:
а - псевдогипертрофия мыщц; б - мышечный валик при миатонической
реакции; в - невозможность расслабить кисти при повторных движениях

Рис. 6.15.

Рис. 6.16. Миотонические феномены у ребенка с миотонией Томсена
кулак и затем быстро его разжать (рис. 6.14-6.16). При этом возникает определенная временная задержка, прежде чем кисть полностью раскроется. При повторном выполнении такого же задания миотонический феномен с каждым разом уменьшается и в конечном итоге исчезает. При врожденной парамиотонии наблюдается обратное явление - нарастание миотонии при повторных движениях (парадоксальная миотония). Перкуссионная миотония проявляется мышечным сокращением после механической стимуляции (быстрый и энергичный удар молоточка по мышце). Этот феномен может наблюдаться в любой мышце, однако наиболее впечатляюще он выглядит при ударе по мышцам тенара: возникает быстрое сгибание и приведение к ладони большого пальца, которое продолжается несколько секунд. При перкуссии крупных мышц возникают симптомы «валика» и «ровика»; при поперечной перкуссии языка образуется «перетяжка» или «ямка» языка. Электромиографическая миотония регистрируется при введении в мышцу игольчатого

Рис. 6.17. ЭМГ при миотонии, «гул пикирующего бомбардировщика»

Рис. 6.18. Миотония Томсена у ребенка. «Геркулесовы мускулы»
электрода. Активное напряжение мышцы или ее перкуссия вызывает появление высокочастотных повторяющихся разрядов, которые вначале увеличиваются по частоте (от 100 до 150 Гц) и амплитуде, а затем уменьшаются. Общая продолжительность таких разрядов около 500 мс, а звуковой эквивалент напоминает гул пикирующего бомбардировщика (рис. 6.17).
Феномен миотонии является важнейшим симптомов нескольких гетерогенных наследственных заболеваний (рис. 6.18, 6.19).
Миотоническая дистрофия, или болезнь Россолимо-Куршмана- Штейнерта-Баттена, является мультисистемным заболеванием, которое наследуется по аутосомнодоминантному типу с вариабельной пенетрантностью патологического гена. Этиология болезни связана с нестабильностью участка ДНК хромосомы 19, что выражается в патологической его амплификации (повторяемости). В результате увеличивается число копий этого гена от 50 до нескольких тысяч. Миотоническая дистрофия с полным правом может быть отнесена к классу так называемых болезней экспансии нуклеотидных триплетов. Число повторов возрастает в последующих поколениях и коррелирует с более тяжелым течением заболевания (феномен антиципации). Количество повторов у ребенка

Рис. 6.19. Миотония Томсена у взрослого больного

Рис. 6.20. Миотония Россолимо- Куршмана-Штейнерта-Баттена. Типичный вид больного
при наследовании болезни от матери возрастает в значительно большей степени, чем при наследовании от отца. У матери с 100 тринуклеотидными повторами риск рождения ребенка с 400 повторами выше 90%.
Болезнь представляет собой самый распространенный вид мышечной дистрофии, дебютирующей у взрослых. Встречаемость заболевания - 3-5 случаев на 100 000 населения. Оба пола поражаются с одинаковой частотой. Первые симптомы обычно появляются у подростков. В развернутых стадиях отмечаются миотония, слабость лицевой мускулатуры и дистальных отделов конечностей, катаракты, лобное облысение, множественная эндокринопатия. Атрофии мимических мышц настолько стереотипны по виду, что все больные выглядят похожими: лицо удлиненное и утонченное вследствие слабости височных и жевательных мышц; шея тонкая («лебединая») из-за атрофии грудиноключично-сосцевидных мышц; веки и углы рта опущены, нижняя половина лица провисает, что делает выражение лица печальным. Атрофии конечностей наиболее выражены в дистальных отделах: предплечьях и перонеальных мышцах (рис. 6.20, 6.21). Отмечается дисфагия, обусловленная поражением мышц глотки и гладких мышц пищевода. Сухожильные рефлексы снижаются и исчезают.

На поздних стадиях заболевания развиваются атрофии мелких мышц кистей. Больные жалуются на напряжение мускулатуры, затруднения при движениях из-за скованности. Миотония нарастает при охлаждении. В целом миотонические феномены не столь выражены, как при врожденной миотонии. Врач может выявить миотонический синдром при расспросе и подтвердить при осмотре. Например, при рукопожатии пациенту с миотонической дистрофией не удается сразу разжать кисть. Экстраневральные симптомы мио- тонической дистрофии - катаракта, лобная алопеция или эндокринные расстройства - возникают еще до клинически значимых симптомов миотонии. Часто регистрируются изменения на ЭКГ. В поздних стадиях может развиваться тяжелая кардиомиопатия с поперечной блокадой, приступами Адамса-Стокса-Морганьи и сердечной недостаточностью. Нарушается перистальтика кишечника, развивается мегаколон. Парез диафрагмы и межреберных мышц приводит к гиповентиляции и рецидивирующим бронхолегочным инфекциям. Эндокринные нарушения включают тестикулярную атрофию, бесплодие у женщин, гиперинсулинизм, сахарный диабет, атрофию надпочечников и нарушение секреции гормона роста. Нередко развиваются гиперсомния и обструктивные апноэ во сне, психические нарушения вплоть до выраженной деменции.
Диагностика базируется на характерных клинических проявлениях и семейном анамнезе. ЭМГ выявляет миотонические феномены, миопатические потенциалы и небольшие признаки денервации. Активность КФК чаще всего соответствует норме. Необходимости в мышечной биопсии для подтверждения диагноза нет. ДНК-анализ обнаруживает увеличение числа тринуклеотидных повторов; он может быть использован для выявления асимптомных пациентов и проведения пренатальной диагностики.
Лечение. Симптомы миотонии ослабевают при назначении препара- тов - мембраностабилизаторов: хинидина, новокаинамида, фенитоина
Рис. 6.21. Миотония Россолимо- Куршмана-Штейнерта-Баттена. «Ле- бединая» шея из-за атрофии грудиноключично-сосцевидной мышцы. Атрофия мышц разгибателей предплечий, перонеальных групп мышц, что приводит к появлению петушиной походки
(дифенина) и карбамазепина (финлепсина). Необходимо учитывать, что миотония сама по себе не инвалидизирует больного и не требует постоянной лекарственной терапии. К сожалению, лечение нарастающей мышечной слабости пока неэффективно. Больные часто относятся к лечению негативно; плохо переносят наркоз, который может осложниться развитием злокачественной гипертермии.
Врожденная миотоническая дистрофия. У матери с миотонической дистрофией вероятность рождения ребенка с врожденной формой болезни составляет 1:4, а если болен отец - 1:12. Основные признаки патологии внутриутробного периода при врожденной форме - снижение двигательной активности плода и многоводие. Преждевременно рождаются 50% детей. Роды могут быть затяжными из-за неадекватного сокращения матки, и часто возникает необходимость наложения акушерских щипцов. У некоторых новорожденных столь грубо страдает функция диафрагмы и межреберных мышц, что они вообще не способны к самостоятельному дыханию. В отсутствие немедленной интубации и ИВЛ многие из них сразу умирают. Наиболее заметные клинические симптомы у новорожденных: лицевая диплегия, при которой рот необычно заострен, а форма верхней губы напоминает перевернутую латинскую букву «V»; генерализованная мышечная гипотония; деформация суставов, варьирующая от двусторонней косолапости до распространенного артрогрипоза; дисфункция желудочно-кишечного тракта в виде пареза мышц желудка, нарушения глотания и аспирации. Слабость наиболее выражена в прок- симальных отделах конечностей. Сухожильные рефлексы отсутствуют. Миотонические феномены не вызываются перкуссией мышц и могут не определяться при ЭМГ. Неонатальная смертность достигает 16% и часто обусловлена кардиомиопатией. У выживших детей мышечная сила, как правило, нарастает, и процессы кормления и дыхания нормализуются в течение 1-го мес жизни.
Отдаленный прогноз неблагоприятен: у всех детей обнаруживаются задержка психического развития и выраженные клинические симптомы миотонической дистрофии. Диагностика требует постановки диагноза миотонической дистрофии у матери, у которой обычно находят множественные клинические признаки заболевания и миотонические ЭМГ-феномены.
Диагноз у матери и ребенка может быть уточнен после проведения амплификации участка ДНК хромосомы 19. Члены семьи входят в группу риска и в дальнейшем проходят генетическое тестирование для установления носительства.
Неотложная помощь новорожденному заключается в немедленной интубации и ИВЛ. Функция желудочно-кишечного тракта нормали- зуется при назначении церукала (метоклопрамида). Тугоподвижность суставов уменьшается при использовании физических методов терапии и иммобилизации.
Врожденная миотония - наследственное заболевание, характеризующееся скованностью и истинной гипертрофией мышц. В 19% семей прослеживается аутосомно-доминантное наследование (болезнь Томсена), реже - аутосомно-рецессивное наследование (болезнь Беккера). Большинство случаев носит спорадический характер. В целом у больных с аутосомно-рецессивной формой заболевание начинается позже и протекает с более тяжелыми миотоническими расстройствами, чем с аутосомно-доминантной. Однако симптомы обеих форм одинаковы, поэтому невозможно сделать вывод о типе наследования исключительно по клиническим критериям (см. рис. 6.18, 6.19).
Патологический ген и доминантной, и рецессивной форм врожденной миотонии картирован на длинном плече хромосомы 7, где расположен ген каналов ионов хлора.
Аутосомно-доминантная форма обычно дебютирует в грудном возрасте с изменения голоса при плаче; ребенок начинает задыхаться, а после плача лицо очень медленно расслабляется. Заболевание протекает легко. В зрелом возрасте миотония может приводить к генерализованной мышечной гипертрофии (атлетизму), но и в детстве мышцы имеют вид «геркулесовых мускулов». Иногда вовлекаются мышцы языка, лица и жевательные мышцы. Скованность мышц не сопровождается болью; она нарастает при пребывании больного на холоде. Выявляются перкуссионные миотонические симптомы. Мышечная масса, сила сокращений и сухожильные рефлексы - в норме. Сразу после отдыха мышцы остаются скованными, а движения - затрудненными. Однако после активизации скованность исчезает, восстанавливается обычный объем движений.
Диагностика. Диагноз подтверждается с помощью ЭМГ-исследования. Частота повторных мышечных осцилляций варьирует от 20 до 80 циклов в секунду от момента первоначального введения иглы в мышцу и до начала произвольного сокращения. Амплитуда и частота потенциалов прибывают и убывают, что сопровождается характерным звуком - «гулом пикирующего бомбардировщика». Признаки мышечной дистрофии отсутствуют. Уровень КФК в норме. В образцах мышечных биоптатов обнаруживается гипертрофия мышечных волокон.
Лечение. Миотония не всегда требует лечения, а лекарства недо- статочно эффективны. Иногда можно уменьшить скованность при назначении фенитоина (дифенина) или препаратов карбамазепина (финлепсина), которые дают в средних противосудорожных дозах. Новокаинамид назначают в начальной дозе 200 мг 2 раза в день, а затем ее постепенно повышают до 400 мг 3 раза в день. Препарат уменьшает мышечную скованность у детей с рецессивной формой болезни. Для некоторых больных эффективен диакарб (ацетазоламид). В тяжелых случаях показан короткий курс кортикостероидов. Полезны антагонисты кальция (нифедипин по 10-20 мг 3 раза в день), а также дизопирамид по 100-200 мг 3 раза в день. Необходимо учитывать, что сукцинилхолин, верошпирон, калий, антигиперлипидемические средства и β-адреноблокаторы способны усиливать миотонический синдром.
Ремиттирующая миотония (миотония, усиливающаяся при избытке калия) - аутосомно-доминантный синдром, связанный с мутацией гена натриевых каналов. Ген картирован на хромосоме 17. Клинические проявления схожи с врожденной миотонией. Дебют мышечной скованности обычно наблюдается после 10 лет и может быть спровоцирован общей анестезией. Миотонические феномены генерализованы, вовлекают туловище, конечности, глазодвигательные мышцы. Тяжесть миотонии день ото дня варьирует и уменьшается при согревании. Ухудшение состояния может наблюдаться после интенсивной физической нагрузки или приема большого количества калия с пищей.
Диагностика. ЭМГ-исследование выявляет миотонические феномены. В мышечных биоптатах патологии нет. Возможен ДНК-анализ мутантного гена, кодирующего α-субъединицу натриевого канала.
Лечение. Скованность при ремиттирующей миотонии может предот- вратить мексилетин - препарат, сходный по структуре с лидокаином; как и при других каналопатиях, может быть эффективен диакарб (ацетазоламид).
6.6. Периодические параличи
Периодические параличи, или пароксизмальная миоплегия, - объединяющий термин для группы каналопатий, редких наслед- ственных заболеваний, которые характеризуются приступами вялого паралича скелетных мышц вследствие патологии ионных каналов. Параличи подразделяют в зависимости от уровня калия в крови: гиперкалиемический (болезнь Гамсторп), гипокалиемический и нормокалиемический. Кроме того, периодический паралич может
быть первичным (генетически детерминированным) или вторичным. Вторичный гипокалиемический периодический паралич обусловлен потерей калия с мочой или его избыточным выведением из желудочнокишечного тракта. «Мочевые» потери калия связаны с первичным гиперальдостеронизмом, интоксикацией солодкой (лакричником), терапией амфотерицином Б и некоторыми почечными тубулярными дефектами. «Желудочно-кишечные» потери калия наиболее часто наблюдаются при тяжелой хронической диарее, длительном зондовом кормлении и гастрофистуле. Калий теряется у подростков с нервной анорексией, которые злоупотребляют диуретиками или искусственно вызывают у себя рвоту, чтобы «похудеть». Гипокалиемический периодический паралич осложняет тиреотоксикоз. Вторичный гиперкалиемический периодический паралич может быть обусловлен почечной или надпочечниковой недостаточностью.
Семейный гиперкалиемический паралич наследуется по аутосомнодоминантному типу с высокой пенетрантностью. Мутация расположена в гене натриевых каналов.
Клиническая картина. Дебют приступов мышечной слабости относится к раннему детскому и даже грудному возрасту. Приступы слабости возникают после интенсивной физической нагрузки. Перед приступом бывают чувствительные нарушения - парестезии в области лица, верхних и нижних конечностей, ощущение тяжести в спине. Изредка больной может замедлить развитие паралича ходьбой или переходом с места на место. У детей грудного и раннего возраста приступы выражаются внезапной потерей мышечного тонуса: они падают и не могут двигаться. У детей старшего возраста и взрослых могут наблюдаться как приступы средней тяжести (продолжаются менее часа и не приводят к глубокому параличу), так и тяжелые приступы (до нескольких часов). После нескольких тяжелых атак может оставаться некоторая остаточная мышечная слабость. Симптомы миотонии у больных с гиперкалиемическим параличом выражены умеренно и могут усиливаться при охлаждении. Характерна миотония век, языка, мышц предплечья и большого пальца.
Диагностика. Во время приступа содержание калия в крови обычно превышает 5 ммоль/л. Пероральный прием хлорида калия сразу после физической нагрузки немедленно провоцирует приступ слабости, на протяжении которого мышцы не реагируют на электрические стимулы.
Лечение. Острые приступы редко требуют лечения, поскольку они кратковременны. При развернутом приступе может помочь внутривен-
ное вливание 40% раствора глюкозы (до 40 мл) или 10% раствора глюконата кальция (до 20 мл). Ежедневный прием диакарба (ацетазоламида) предотвращает повторные приступы, механизм превентивного действия этого препарата при гиперкалиемическом и гипокалиемическом параличе неизвестен. Следует избегать приема пищи, богатой калием, увеличить в дневном рационе количество углеводов и поваренной соли.
Семейный гипокалиемический паралич наследуется по аутосомнодоминантному типу. Пенетрантность гена у женщин снижена. Мутация находится на длинном плече хромосомы 7, в гене кальциевых каналов. У 60% больных симптомы возникают до 16 лет, у остальных - до 20 лет жизни. Вначале приступы слабости нечастые, но затем бывают до нескольких раз в неделю. Приступы провоцируют: отдых после физической нагрузки (часто приступы наблюдаются ранним утром), обильный прием углеводистой пищи, избыток поваренной соли в рационе, эмоциональный стресс, прием алкоголя, переохлаждение; у женщин - менструации. До и во время приступа у больного могут отмечаться жажда и олигурия, болезненные ощущения в проксимальных группах мышц, затем развивается общая слабость. Иногда наблюдается тотальный паралич, при котором больной не в состоянии даже поднять голову. Слабость лицевых мышц бывает редко, движения глаз всегда сохранены. Дыхательная недостаточность не развивается. Большинство приступов длится от 6 до 12 ч, а некоторые - в течение всего дня (так называемый миоплегический статус). Мышечная сила быстро восстанавливается, но после нескольких тяжелых приступов могут отмечаться усталость, похудание, особенно проксимальных отделов конечностей, угнетение сухожильных рефлексов. Типичны вегетативные расстройства: гиперемия кожи, гипергидроз, лабильность пульса и артериального давления. Вне приступов мышечной слабости у больных отсутствуют симптомы нервно-мышечной патологии.
Диагностика. Во время приступа уровень калия в крови может снизиться до 1,5 ммоль/л, чему соответствуют изменения ЭКГ: брадикардия, уплощение зубца Т, увеличение интервалов Р-Q и Q-Т. Мышцы не сокращаются в ответ на электрические стимулы. С диагностической целью приступ можно спровоцировать приемом глюкозы в дозе 2 г/кг и одновременным подкожным введением 10-20 ЕД инсулина: приступ паралича развивается через 2-3 ч.
Лечение. Острые приступы у больных с адекватной функцией почек купируют повторными приемами калия в дозе от 5 до 10 г.
Такая же доза, принимаемая ежедневно, рекомендуется для профилактики их возникновения. У детей младшего возраста доза меньше. Ежедневный прием диакарба (ацетазоламида) оказывает благоприят- ное действие, предупреждая приступы во многих случаях. Он малотоксичен и обычно хорошо переносится даже при длительном применении. Следует снизить калорийность суточного рациона за счет углеводов и уменьшить количество поваренной соли. В то же время показаны продукты, богатые калием: сухофрукты, курага, чернослив, молочные продукты, картофель.
Семейный нормокалиемический паралич. В некоторых семьях наблюдаются случаи аутосомно-доминантного периодического паралича при нормальном уровне калия в крови. Это вариант гиперкалиемического периодического паралича с нарушением притока калия в кровь, когда невозможно оценить его истинное содержание в тканях. Миоплегия длится от нескольких дней до 2-3 нед. Темп нарастания и уменьшения мышечной слабости обычно медленный. Сухожильные рефлексы во время приступов исчезают. У части больных наблюдается гипертрофия отдельных мышечных групп. Приступы провоцируются отдыхом после интенсивной физической нагрузки, приемом алкоголя, охлаждением. Прием хлорида калия может спровоцировать приступ паралича, тогда как употребление 8-10 г поваренной соли ежедневно позволяет их избежать.
6.7. Миастения
Миастения (myastenia gravis) - аутоиммунное нервно-мышечное заболевание, клинически характеризующееся патологической слабостью и утомляемостью произвольной мускулатуры и связанное с повреждением ацетилхолиновых рецепторов (АХ-Р) постсинаптической мембраны поперечнополосатых мышц специфическими ком- плементфиксирующими антителами (АТ).
Распространенность миастении составляет 0,5-5 случаев на 100 000 населения во всех популяциях. Дети и подростки до 17 лет составляют 9-15% числа больных миастенией. Средний возраст начала болезни - 7,2 года. Дебют миастении возможен в любом возрасте. Описаны врожденные формы. Женщины болеют в 3 раза чаще, чем мужчины.
Этиология. Мультифакториальное заболевание, при котором определенное значение имеет наследственная предрасположенность, обусловленная иммунологическим дефектом и связанная с анти-
генами гистосовместимости B8 системы HLA. Причиной миастении могут быть вирусное поражение вилочковой железы, вследствие чего она начинает продуцировать Т-лимфоциты с измененными мембранными структурами; опухоль тимуса; в редких случаях - первичное поражение головного мозга различной этиологии.
Основой патогенеза миастении является аутоиммунная реакция на ацетилхолинэстеразные рецепторы (АХ-Р) скелетных мышц. Уровень антител к АХ-Р в крови больных коррелирует со степенью тяжести болезни. Антитела к АХ-Р блокируют нервно-мышечную проводимость, поскольку разрушают АХ, уменьшают скорость его восстановления, необратимо изменяя рецепторы постсинаптической мембраны.
Патологическая анатомия. Происходят дистрофические изменения терминалей аксонов, синаптических щелей и постсинаптических структур, в них откладываются иммуноглобулины и комплемент. В мышцах наблюдается умеренная дегенеративная атрофия, реже некроз волокон в сочетании с нерезко выраженной лимфоидной инфильтрацией и плазморрагией. У 70-90% больных выявляется патология вилочковой железы (гиперплазия зародышевых фолликулов, лимфоэпителиальные тимомы). В редких случаях отмечаются миокардит, тиреоидит, очаговые скопления лимфоцитов в различных органах.
Клиническая классификация миастении (по Б.М. Гехту).
1. Степень генерализации двигательных расстройств:
1) генерализованная;
2) локальная:
а) глазная,
б) бульбарная,
в) скелетная.
2. Степень тяжести двигательных нарушений:
1) легкая;
2) средняя;
3) тяжелая.
3. Течение миастенического процесса:
1) ремиттирующее (миастенические эпизоды);
2) непрогрессирующее (миастеническое состояние);
3) прогрессирующее;
4) злокачественное.
4. Степень компенсации двигательных расстройств под влиянием антихолинэстеразных препаратов:
1) полная (вплоть до восстановления работоспособности);
2) неполная (восстанавливается способность к самообслуживанию);
3) плохая (больные нуждаются в постороннем уходе). Клиническая картина. Миастения характеризуется патологической
утомляемостью и слабостью поперечнополосатых мышц. Больным трудно подниматься по лестнице, ходить, долго находиться в одной позе, носить тяжести.
Наиболее часто поражаются глазодвигательные, мимические, жевательные мышцы, а также мышцы глотки, гортани, языка. Поражение наружных мышц глаза во время первого осмотра выявляется у 40-50% больных, а по мере развития болезни - у 90-95%. Птоз может быть односторонним, причем возникает то с одной, то с другой стороны. Утром и после отдыха птоз меньше, нарастает при общей или зрительной нагрузке, к вечеру. При осмотре спровоцировать усиление птоза можно, попросив пациента несколько раз зажмуриться или присесть. Глазодвигательные нарушения асимметричны, изменчивы при нагрузке и не соответствуют зонам иннервации глазодвигательных нервов. За счет мышечной слабости возникает нистагм в крайних отведениях. Диплопия усиливается при зрительной и физической нагрузке, ярком свете, во второй половине дня (особенно при просмотре телевизора), более выражена при взгляде вдаль, уменьшается после отдыха с закрытыми глазами и по утрам (рис. 6.22).
Слабость жевательных и височных мышц приводит к утомлению при жевании иногда до отвисания нижней челюсти, больные во время еды поддерживают челюсть и помогают себе при жевании рукой. Важным сим- птомом является слабость мышц лица. Она более выражена в верхней половине лица (в круговых мышцах глаз), усиливается при повторном зажмуривании и общей физической нагрузке. Пациенту трудно надуть щеки, возникает «поперечная» улыбка вследствие слабости круговой мышцы рта. Также отмечается слабость жевательных и височных мышц.

Рис. 6.22. Слабость глазных мышц при миастении
Поражение бульбарных мышц (мягкого нёба, глотки и верхних мышц пищевода), приводящее к дисфагии и дизартрии, развивает- ся у 40% больных. Оно усиливается при речевой, общей физической нагрузке, во время еды и уменьшается после отдыха. Нарушается глотание (пациент поперхивается при еде, жидкая пища попадает в носовые ходы). Речь становится гнусавой, может отмечаться охриплость голоса или нарушения модуляции, похожие на заикание. При тяжелой дизартрии больной не может глотать и говорить.
Слабость мышц шеи и туловища больше характерна для больных пожилого возраста. Слабость мышц спины проявляется нарушением осанки. За счет слабости задней группы мышц шеи возникает затруднение при подъеме головы в положении на спине или при разгибании шеи в вертикальном положении. Если миастения дебютирует со слабости мышц туловища, в дальнейшем развиваются бульбарные и дыхательные нарушения.
Жалобы на одышку при вдохе обусловлены слабостью диафрагмы или межреберных мышц. Ослабление кашлевого толчка приводит к скоплению густой мокроты, вязкой слюны, которую невозможно сплюнуть или проглотить.
Ослаблены мышцы конечностей, особенно проксимальных, шеи, туловища. При осмотре выявляются мышечные атрофии, снижение мышечного тонуса, лабильность сухожильных и надкостничных рефлексов. Слабость мышц конечностей может быть изолированной (без других симптомов миастении) или сочетаться со слабостью других групп мышц. Типична слабость проксимальных мышц-разгибателей. Наиболее часто поражаются дельтовидная мышца, трехглавая мышца плеча, подвздошная мышца.
Кроме двигательных расстройств, миастения сопровождается различными вегетативными и эндокринными нарушениями (гипо- и гипертиреоз, гипокортицизм и др.). Миастению характеризуют динамичность мышечной слабости в течение суток, усиление ее после нагрузки, обратимость или уменьшение слабости после отдыха. Ухудшение состояния провоцируют физическая нагрузка, отрицательные эмоции, менструация, инфекции, повышение температуры окружающей среды, а улучшают - ночной сон, отдых. Патогномонично уменьшение утомляемости после введения антихолинэстеразных препаратов (АХП).
Течение болезни чаще всего прогрессирующее, с ремиссиями или прогредиентное без ремиссий. При злокачественном течении буль- барные и дыхательные расстройства развиваются в течение первых недель заболевания. Миастения чаще дебютирует после ОРВИ или
стресса, одним симптомом (преходящий птоз, бульбарный парез и др.). Состояние больных с миастенией могут осложнять миастенические кризы или холинергические кризы.
Миастенический криз развивается вследствие декомпенсации миастении или недостаточной дозировки АХП; может провоцироваться бронхолегочной инфекцией. При этом происходит резкое ухудшение состояния с нарушением витальных функций. Дифференцировать миастенический криз от других тяжелых состояний, сопровождающихся респираторными расстройствами, можно по наличию асимметричного наружного офтальмопареза, птоза, бульбарного синдрома, гипомимии, слабости мышц конечностей и шеи, уменьшающихся в ответ на введение АХЭ препаратов (табл. 10).
Холинергический криз развивается при избыточной дозе АХЭ препаратов.
Таблица 10. Дифференциальная диагностика миастенического и холинергического кризов

Смешанные (миастенический + холинергический) кризы встречаются у больных миастенией при неправильном приеме и/или изначально узком диапазоне терапевтических доз АХЭП, а также на фоне состояний, вызывающих общую или мышечную слабость различного генеза (интеркуррентные инфекции, соматические, гормональные нарушения, прием препаратов, влияющих на сократительную функцию произвольных мышц, и др.).
Прогноз зависит от клинической формы и проводимого лечения. Возможны практические выздоровления (примерно у 1 / 3 больных), значительное улучшение, инвалидность, летальные исходы, особенно при тимоме. Основными симптомами, угрожающими жизни больного, являются слабость мышц гортани и дыхательных мышц. Причины смерти при миастении: дыхательная недостаточность, аспирационная пневмония, побочные эффекты кортикостероидов и цитотоксических препаратов.
Диагностика включает сбор анамнеза, клинический осмотр, пробу с АХЭ-препаратами (прозерином, тензилоном, калимином), электромиографию, иммунологическое исследование, исследование вилочковой железы, морфологическое исследование мышечного биоптата, динамическое наблюдение.
Клинический осмотр включает исследование общего неврологического статуса и оценку силы произвольных мышц лица, шеи, туловища и конечностей до и после нагрузки. Мышечная сила оценивается от 0 до 5 баллов, где 0 - отсутствие силы, 5 - нормальная сила с учетом возраста и пола. Также выявляют синдром патологической мышечной утомляемости (нарастание симптомов после нагрузки) при отсутствии симптомов поражения центральной нервной системы.
Диагностические критерии
1. Птоз (односторонний, двусторонний, асимметричный, симметричный): появление или усиление птоза после долгого смотрения вверх или после быстрого многократного открывания или закрывания глаз.
2. Слабость жевательных мышц:
Недостаточное сопротивление насильственному закрытию нижней челюсти;
Пальпация височных мышц при жевании обнаруживает их слабое сокращение;
Больные не в состоянии плотно сомкнуть веки или оказать сопротивление пассивному открыванию глаз;
Больные не могут надуть щеки при надавливании на них.
3. Слабость мышц гортани и нёба выявляется, если:
Нёбо малоподвижно, рвотный рефлекс снижен или отсутствует;
Трудно глотать жидкую пищу.
4. Слабость мышц языка выявляется при надавливании языком на палец врача через щеку.
5. При выраженной слабости мышц шеи «голова свешивается».
6. Прозериновая проба с оценкой силы и утомляемости мышц проводится до подкожного введения 0,05% раствора прозерина в разовой возрастной дозировке и через 30-40 мин после него. Проба считается положительной, если мышечная сила нарастает. Различают:
Резкоположительную пробу, когда исчезают все миастенические симптомы;
Положительную пробу - остаются только отдельные симптомы;
Слабоположительную пробу, при которой уменьшается выраженность миастенических симптомов;
Сомнительную прозериновую пробу - степень выраженности проявлений миастении изменяется незначительно;
Отрицательную прозериновую пробу - клиническая симптоматика не изменяется после введения прозерина.
Подтверждением диагноза миастении считается наличие одного из первых трех вариантов прозериновой пробы.
Проводят ЭМГ наиболее ослабленных мышц для выявления особенностей нарушений нервно-мышечной передачи (мышца, отводящая мизинец, в двубрюшной мышце дна рта). Исследование проводят на фоне отмены АХЭП в течение суток, сразу после физической нагрузки и через 2 мин после нагрузки. Большое значение имеет обратимость ЭМГ-феноменов на фоне АХЭП - нарастание амплитуды М-ответа. При электромиографии отмечается уменьшение амплитуды второго потенциала действия мышц (в норме оба потенциала равны) в ответ на стимуляцию нерва спаренными импульсами с промежутком 0,1-0,7 с. При миастении снижение амплитуды потенциалов при постоянной стимуляции нерва сменяется фазой плато или повышением амплитуды, а при других заболеваниях происходит неуклонное снижение амплитуды ответа. При регистрации активности отдельных мышечных волокон часто выявляются характерные признаки поражения нервно-мышечных синапсов. В 95% случаев на ЭМГ обнаруживают патогномоничные изменения.
Для исключения опухоли или гиперплазии вилочковой железы, которая развивается у 75% больных с миастенией, проводят компьютерную томографию средостения, радионуклидное сканирование.
Иммунологическое исследование выявляет наличие антител к холинорецепторам у 50% больных с глазной формой миастении и у 80-90% больных с генерализованной формой. При тимоме также выявляются антигены к скелетным мышцам.
Иммунологическое исследование (ИФА, РИА) - количественный метод определения антител к АХР в сыворотке крови больных миастенией, который позволяет с вероятностью до 80% подтвердить диагноз.
Дифференциальный диагноз проводят с состояниями, ведущим симптомом которых является мышечная слабость:
Миастенические синдромы (ботулизм, отравление антибиотиками из группы аминогликозидов, болезнь Иценко-Кушинга, болезнь Аддисона, гипо- и гипертиреоз, полиомиозит);
Рассеянный склероз, нейроинфекции (энцефалит, полиневропатии, энцефаломиелополирадикулоневриты): у больных офтальмопарез сопровождается гипорефлексией, атаксией, нарушением чувствительности, изменением ЦСЖ;
Боковый амиотрофический склероз: слабость постоянна, отмечаются атрофия, фасцикуляции, повышение сухожильных рефлексов, симптом Бабинского;
Глазная форма миопатии: характерны птоз и симметричное ограничение движений глазных яблок; легкая слабость мышц глотки, шеи, конечностей и лица;
Митохондриальные миопатии;
Нейроэндокринные синдромы;
Другие заболевания ЦНС (опухоли, сосудистые заболевания головного и спинного мозга): характерны нарушения рефлексов, проводниковые расстройства;
Астеноневротические реакции, синдром хронической усталости и др.
Лечение. Общие принципы:
1. При генерализованной форме больного госпитализируют и ограничивают физическую нагрузку до подбора антихолинес- теразной терапии.
2. Противопоказаны средства, блокирующие нервно-мышечную передачу, а также оказывающие угнетающее действие на ЦНС,
и особенно - на дыхательный центр (хинин, хинидин, пропранолол, лидокаин, аминогликозиды, полимиксин, морфин, барбитураты, транквилизаторы). 3. Задачи лечения зависят от тяжести заболевания. Антихолинестеразные препараты (АХЭП) - препараты выбора при миастении, тормозят разрушение ацетилхолина и способствуют его накоплению в синаптической щели, воздействуя на холинергические синапсы, не проникают через ГЭБ (табл. 11). Побочные эффекты обусловлены одновременным влиянием на вегетативные холинергические синапсы и зависят от дозы и тонуса ВНС. Их можно уменьшить, если принимать ингибиторы АХЭ чаще, но в меньших дозах и во время еды, что замедляет всасывание. В некоторых ситуациях (менструация, инфекции, ремиссия) чувствительность к АХЭП повышается, и их дозу уменьшают. Больных обучают регулировать дозу самостоятельно. Относительными противопоказаниями к применению АХЭП являются бронхиальная астма, тяжелый атеросклероз, ИБС, эпилепсия.
Таблица 11. Антихолинергические препараты
Препараты | Время действия | Области применения |
Прозерин (неостигмин) | Начало действия через 20-40 мин, дли- тельность 2-4 ч | Применяется преимущественно для проведения медикаментозных проб и в острых состояниях |
Калимин 60 Н, калимин-форте (пиридостигми- на бромид) | Начало через 45 мин, действует 4-8 ч Интервал между приемами 5-5,5 ч. | Наиболее широко применяется, хорошо переносится, эффективен при всех формах, в том числе бульбарных. Калимин форте (парентеральный) - при нарушении витальных функций и при стойком бульбарном параличе. При переводе больных на парентеральное введение препаратов учитывается, что 1 таблетка калимина (60 мг) равноценна 1 мл 0,05% раствора прозерина |
Вспомогательная терапия: препараты калия (пролонгируют действие АХЭП); диета, богатая калием (печеный картофель, курага, бананы и др.); калийсберегающие препараты (верошпирон); хлорид калия 3,0 г/сут в растворах, порошках, таблетках с целью предупреждения передозировки АХЭП; препараты кальция; тонизирующие средства (экстракты элеутерококка, родиолы, левзеи, пантокрин); поливитамины, эуфиллин (блокатор фосфодиэстеразы, увеличивающий содержание цАМФ в пресинаптической мембране), анаболики (рибоксин, ретаболил).
Патогенетическая терапия - тимэктомия. Эффективность - 70-90%, возможны ремиссии. Показаниями к оперативному лечению являются:
а) злокачественные формы миастении;
б) прогрессирующая форма миастении;
в) миастеническое состояние в зависимости от степени выраженности дефекта.
Противопоказания к тимэктомии:
а) тяжелые декомпенсированные соматические заболевания;
б) пожилой возраст.
Предоперационная подготовка включает общеукрепляющую терапию, плазмаферез, по показаниям - глюкокортикоиды, лучевую терапию (противопоказана детям и подросткам).
Глюкокортикоиды (преднизолон, дексаметазон) показаны при неэф- фективности других методов. Их назначают ежедневно или через день, по 60-150 мг/сут (1-1,5 мг/кг/сут) утром, сразу после завтрака, через день; при выраженном обострении ежедневно (до компенсации витальных нарушений), через 5-7 дней (до терапевтического эффекта) переходят на схему через день. Поддерживающая доза - через день 20-30 мг в сутки, принимается в течение нескольких месяцев. Примерно у 75% больных кортикостероидная терапия приводит к существенному улучшению. После стабильного улучшения дозу кортикостероидов медленно (в течение нескольких месяцев) снижают до поддерживающей (5-15 мг ежедневно или 10-30 мг через день). Иногда удается полностью отменить кортикостероиды. Чтобы избежать первоначального ухудшения, лечение можно начинать с низких доз (25 мг преднизолона через день) с постепенным увеличением дозы на 12,5 мг в каждый третий прием, пока суточная доза не достигнет 100 мг или не будет получен хороший эффект. Улучшение отмечается спустя 6-7 нед лечения. Дозу в этих случаях начинают снижать не раньше чем через 3 мес после первого приема.
Плазмаферез назначают при обострениях, миастенических кризах, предоперационной подготовке, неэффективности кортикостероидной терапии. Проводится 3-5 сеансов через день, затем 2-3 раза в неделю. Плазмаферез проводят с заменой плазмы или использованием белков-заменителей. Гемосорбция и энтеросорбция проводятся у больных с генерализованной формой миастении с целью выведения антител, а при смешанных кризах и неэффективности массивной медикаментозной терапии - с целью детоксикации.
Цитостатики (азатиоприн, циклофосфан и циклоспорин) назначают под контролем анализов крови. Препараты иммуноглобулина G (в/в 0,4 г/кг/сут ежедневно в течение 5 дней; или 3-5 г на курс) эффективны при интеркуррентных инфекциях, во время миастенического или смешанного криза.
Лечение кризов направлено на компенсацию витальных нарушений, купирование обострения и устранение метаболических нарушений. При лечении миастенического криза вводят парентерально АХЭП (калимин-форте по 1-1,5 мл в/в или в/м каждые 4-5 ч или прозерин по 1,5-2 мл каждые 3 ч). ИВЛ с полной отменой АХЭП, назначение иммуносупрессивной терапии на фоне антибактериальных пре- паратов проводят с целью профилактики интеркуррентных инфекций. Отключение от аппарата проводят только после 30 мин самостоятельного дыхания, при компенсации дыхательных расстройств и на фоне калимина-форте на 5-6 ч. Большие дозы глюкокортикоидов назначают по альтернирующей схеме (пульс-терапия - 1000-2000 мг в/в капельно через день) с последующим переводом на пероральный прим. Также стабилизируют сердечно-легочную деятельность. Проводят плазмафе- рез, внутривенные инфузии нормального человеческого иммуноглобулина. Холинергический криз купируют атропином, реактиваторами холинэстеразы (дипироксим); применяют дезинтоксикацию.
Кроме болезненных нарушений, полученных при травмах (например, разрывов и растяжений), нарушения в мышцах могут быть и при отсутствии внешних факторов воздействия. К болезням мышц можно отнести:
Мышечная судорога;
Ревматические заболевания;
Воспаления;
Генетические заболевания;
Нарушение обмена веществ;
Изменение мышечных клеток.
Рассмотрим все заболевание более подробно.
Мышечная судорога
Судорога может появиться в результате обезвоживания организма (эксикоза). В этот момент мышцы сократились и стали твердыми, затем они медленно расслабляются. Судорога может возникнуть ночью или утром. Человек резко начинает ощущать сильную боль в мышце. Судороги наиболее часто встречаются у пожилых людей. Когда на мышцы идет слишком большая нагрузка или питание их нарушается, то появляются затвердения. Мышечные волокна перевоплощаются в мышечную ткань, в которой прощупываются твердые участки в виде узлов. В таких случаях необходимо пить много жидкости, для того чтобы восстановить водно-солевой баланс в организме. Также на помощь приходит массаж. Если боли в мышцах не прекращаются, то необходимо показаться врачу. Лечат затвердение при помощи массажа, витамина Е и теплых ванн.
Ревматические заболевания
Есть очень большое количество заболеваний, который можно отнести к ревматическим.. При этих заболеваниях источник поражения – сама мышца, либо кровеносные сосуды, которые питают мышцу. Появляются боли в бедрах и плечах. Некоторые заболевания ревматического характера (например, дерматомиозиты) поражают мышцы. В данном случае необходимо лечение гормонами – глюкокортикоидами. Они подавляют воспаление, но вызывают побочные эффекты. Поэтому ревматические заболевание пробуют подавить с помощью противовоспалительных лекарств или физиотерапии.
Нарушение гормонального характера
Болезненная слабость мышц в медицине именуется как эндокринная миопатия, которая появляется вследствие усиления функции щитовидной железы или надпочечников. После лечения боли исчезают.
Воспаление мышц
Воспаление мышц называется миозитом. Симптомы данного заболевания такие же, как и при ревматизме, но отличительной чертой является воспаление самих мышц. Для миозитов присущи боли и яркая мышечная слабость. Лечат воспаления мышц точно так же, как и ревматические заболевания.
Недостаток минеральных веществ
Для нормального функционирования мышцам необходимы определенные вещества. При дефиците калия возникает паралич. Особенно это чувствуют молодые люди и детки утром после тяжелого прошлого дня. Лечат с помощью препаратов, содержащих калий. Кроме того, перед сном не стоит много есть и активно заниматься спортом.
Недостаток ферментов
У детей может редко наблюдаться дефицит фермента. Часто встречаются нарушения функций ферментов, которые принимают участие в расщеплении глюкозы и гликогена, которые являются источником энергии для мышц. В результате врожденного дефицита фермента мышцы получают мало энергии вследствие ослабления их работы. Человек с таким диагнозом должен избегать физической нагрузки.
Болезненная усталость мышц
Усталость мышц, которая сопровождается болью, появляется вследствие ацидоза. Для получения энергии при больших нагрузках происходит расщепление глюкозы до молочной кислоты, которая тяжело выводится из организма. Накапливаясь в мышцах, молочная кислота вызывает болезненные ощущения.
Во всем мире спортсмены для предотвращения боли в мышцах, улучшения питания, восстановления и лечения употребляют сок из мангустина.
Необходимо употреблять чистую воду.
Под миозитом подразумевается группа самых различных по этиологии патологических процессов в скелетной мускулатуре. В узком понимании миозит – это воспаление скелетной мускулатуры, то есть той мышечной ткани, которая обеспечивает движение опорно-двигательного аппарата (а не гладкой мускулатуры внутренних органов ). Однако миозит может быть не только воспалительным, но и травматическим или токсическим.Миозит может быть как самостоятельным заболеванием (оссифицирующий миозит ), так и одним из проявлений других патологий (например, туберкулеза ). Очень часто миозит сопутствует аутоиммунным заболеваниям, таким как системная красная волчанка и ревматоидный артрит . Одной из самых тяжелых форм миозита является дерматомиозит или болезнь Вагнера, при котором совместно с мышечной и соединительной тканью поражаются кожные покровы.
Если миозитом поражается несколько групп мышц – то тогда он называется полимиозитом, если же поражается одна мышца – то тогда он носит название локального миозита. Совместно с мышечной тканью могут поражаться кожные покровы (дерматомиозит ), или же нервные волокна (нейромиозит ).
Самым распространенным видом миозита – является шейный миозит, он составляет более половины случаев (50 – 60 процентов ). На втором месте – поясничный миозит, который является самой частой причиной болей в пояснице .
Сегодня миозит считается офисной болезнью. Для представителей «сидячих» профессий риск развития этой патологии значительно выше, чем у представителей «подвижных» профессий. Неудобная и вынужденная поза, например, за компьютером в течение 6 – 8 часов с поддувающим кондиционером за спиной чревата развитием поясничного или шейного миозита.
Некоторые виды миозита считаются профессиональными, например, у скрипачей или пианистов, что обусловлено постоянным напряжением мышц кисти, шеи или спины.
Считается, что различными видами миозита страдают более половина жителей мегаполисов.
Причины миозита
 Условно причины миозита можно разделить на эндогенные (причины, возникшие внутри самого организма
) и экзогенные (причины, возникшие вне организма
).
Условно причины миозита можно разделить на эндогенные (причины, возникшие внутри самого организма
) и экзогенные (причины, возникшие вне организма
).Название «аутоиммунный» отражает патогенез и характер заболевания. При этой патологии происходит выработка самим организмом антител к собственным тканям (в данном случае к соединительной ткани
), на которых фиксирован антиген. Антигеном может быть вирус , бактерия , грибок . При образовании комплекса антиген-антитело запускается каскад воспалительных реакций, с дальнейшим поражением ткани. Как правило, миозит такой этиологии (чаще всего это так называемый ревматический миозит
), имеет подострое или хроническое течение и характеризуется тянущими болями.
Инфекции
Большинство инфекций протекает с развитием миозита. При этом инфекция из основного очага (будь то миндалины или легкие ) распространяется с током крови или лимфы на мышечную ткань. В дальнейшем в мышце (или группе мышц ) развивается воспаление специфического или неспецифического характера.Различают инфекционный гнойный и негнойный миозит. Негнойный миозит развивается в период гриппа, различных респираторных заболеваний, сифилиса , брюшного тифа , туберкулеза. Особой формой негнойного миозита является Борнхольмская болезнь или эпидемическая миалгия. Это острое инфекционное заболевание, вызывающееся энтеровирусом Коксаки, которое поражает преимущественное мышечную систему. Ведущим симптомом этого заболевания являются сильные боли в области живота и грудной клетки на фоне лихорадки.
Гнойные миозиты развиваются на фоне генерализованной гнойной инфекции (чаще всего стафилококковой или стрептококковой ) или остеомиелита. При этом патогенный микроорганизм разносится с током крови к мышцам, где в дальнейшем формируются локализованные гнойные очаги. Таким образом, в мышечной ткани образуются скопления гноя, участки некроза и флегмоны. Гнойный миозит является очень тяжелым заболеванием и требует хирургического вмешательства.
Различные интоксикации
Миозит может развиться в результате воздействия на организм различных токсических веществ. Чаще всего токсический миозит наблюдается при алкоголизме , но также встречается при приеме некоторых медикаментов, отравлениях, укусах насекомых .Механизм развития токсического миозита состоит в прямом токсическом воздействии алкоголя, медикамента или яда.
Прямым разрушающим мышцу действием обладают:
- алкоголь;
- антималярийные препараты;
- колхицин;
- кортикостероиды;
- изониазид.
Травмы
На месте травмы происходит разрыв мышечных волокон, с дальнейшим развитием воспалительного отека . В дальнейшем по мере заживления отек заменяется рубцовой тканью, а мышца при этом укорачивается.Также результатом травм может быть развитие так называемого оссифицирующего миозита. При этом в толще мышцы, а именно в области соединительнотканных участков, развиваются участки окостенения.
Постоянное напряжение мышц
Эта причина характерна для профессиональных миозитов. В результате длительного напряжения или неудобного положения мышца напрягается и уплотняется. При этом в ней нарушается процесс питания, так как ток крови в напряженной мышце замедляется. Нарушенное кровообращение как следствие является причиной недостатка кислорода и развития дистрофических процессов в мышце.Переохлаждение
Сквозняки, конечно же, являются самой частой причиной развития миозита. Чаще всего переохлаждению подвержены мышцы спины, поясницы и шеи. При этом в процесс могут вовлекаться не только мышцы, но и нервные волокна.Виды миозита
 Существует две основные формы миозита – локальный миозит и полимиозит. Локальный миозит характеризуется воспалением одной мышцы. При полимиозите воспалительный процесс распространяется на несколько мышц или группы мышц.
Существует две основные формы миозита – локальный миозит и полимиозит. Локальный миозит характеризуется воспалением одной мышцы. При полимиозите воспалительный процесс распространяется на несколько мышц или группы мышц.Областями, в которых чаще возникает миозит, являются:
- поясница;
- руки;
- ноги;
- челюстно-лицевая область.
Миозит шейного отдела
Миозит шейного отдела возникает чаще, чем в других областях тела. При этом появляются боли в области шеи, которые могут распространяться как наверх (к затылку, ушам ), так и вниз между лопатками. Боль может быть настолько сильной, что сковывает движения шеи.
Миозит в области поясницы
Миозит в области поясницы затрагивает поясничные мышцы вдоль позвоночника . Боль менее выражена, чем при шейном миозите, и носит ноющий характер. При пальпации поясничной области отмечается уплотнение мышц и усиление болей. Миозит поясничной области более распространен среди населения пожилого возраста.
Миозит мышц рук и ног
Миозит мышц рук и ног встречается редко в виде локальных форм. Чаще воспаление мышц конечностей наблюдается при полимиозите. Больному сложно передвигать ноги, поднимать руки выше головы. Снижение силы в мышцах сопровождается появлением болей при их напряжении.
Миозит жевательных мышц - нередко наблюдается в челюстно-лицевой области. При данной форме боли возникают или усиливаются при жевании.
Полимиозит встречается чаще, чем локализованные формы миозита.
Полимиозит с признаками дерматита называется дерматомиозитом. Вследствие длительного воспалительного процесса мышцы истончаются и атрофируются.
Полимиозит чаще появляется у людей среднего возраста (30 – 60 лет
). Однако есть отдельная форма полимиозита, которая появляется только у детей от 5 до 15 лет. Женский пол подвержен заболеванию в два раза чаще, чем мужской. Появлению заболевания могут предшествовать различные вирусные инфекции, переохлаждение, снижение иммунитета , большие физические нагрузки и травмы. Болезнь развивается медленно, в течение недель и месяцев. Первым проявлением является усталость и слабость мышц дистальных частей тела (особенно бедренных, плечевых и шейных мышц
). Слабость усиливается и иногда даже переходит в умеренную боль. Все движения затруднены и замедленны. Больным сложно поднимать руки, ходить, вставать со стула или кровати. Появляется дисфагия (затрудненное глотание
), затрудняется дыхание и речь. При дерматомиозите появляются кожные высыпания пурпурного цвета, которые немного возвышаются над кожей. Поражение внутренних органов при полимиозите наблюдается редко.
Нейромиозит
Нейромиозит – это одна из форм полимиозита, характеризующаяся поражением мышечных волокон и нервов, которые расположены в данной области. В большей степени поражаются внутримышечные нервные волокна, однако нередко и дистальные отделы нервов (особенно когда болезнь прогрессирует ). При воспалении мышечные клетки разрушаются и высвобождаются различные вещества, которые токсично влияют на нервные волокна. Также нервные волокна подвержены воздействию Т-лимфоцитов, которые высвобождаются при аутоиммунной реакции. Под действием этих клеток и всех компонентов воспалительной реакции, миелиновая оболочка нерва разрушается. Если процесс не остановлен, то вскоре разрушается и осевой цилиндр нервного волокна.Основными признаками нейромиозита являются:
- парестезии в пораженной зоне (снижение чувствительности );
- гиперестезии (повышение чувствительности );
- выраженные болевые ощущения;
- симптомы натяжения;
- снижение тонуса и силы мышц;
- болезненность в суставах.
Боль при нейромиозите прогрессирует. Вначале она умеренная, затем усиливается при небольших нагрузках. Боль может появиться или усилиться при дыхании, при поворотах и наклонах тела, при движении рук и ног. Постепенно боль появляется даже в состоянии покоя. Болевой синдром сильно выражен, когда поражены дистальные части нервов.
Также важным признаком нейромиозита является симптом натяжения. Пальпация мышц в натянутом напряженном состоянии вызывает болевые ощущения. Обычно нейромиозит сопровождается суставными болями, реже - кожными поражениями.
Полифибромиозит
Полифибромиозит – это другая форма полимиозита, основной особенностью которой является замещение мышечной ткани на соединительную ткань.Вследствие длительного воспалительного процесса в мышечной ткани мышечные клетки разрушаются и фиброзируются (замещаются клетками соединительной ткани ). Другими словами, на месте поврежденной мышечной ткани появляется рубец. Рубцовая ткань уплотняется в виде узелков, которые хорошо чувствуются при прощупывании мышц. При образовании рубцовой ткани нередко формируются спайки между мышцами. Когда рубцы образуются вблизи сухожилий, появляются разные контрактуры и снижается подвижность.
Основными признаками полифибромиозита являются:
- уплотнение пораженных участков мышцы;
- образованием узелков;
- контрактуры и анормальные сокращения мышц;
- уменьшение амплитуды движений, снижение подвижности;
- боли при движении и при пальпации мышц.
Оссифицирующий миозит
Оссифицирующий миозит – очень редкая форма полимиозита, который может развиться после травмы (ушибов, вывихов , переломов , растяжений и разрывов ). Это может быть как результат острой травмы, так и хронического повреждения мышц. Так, например, у наездников при верховой езде постоянно травмируются мышцы бедра, у фехтовальщиков – мышцы груди. Также есть случаи врожденной болезни, которая прогрессирует с возрастом. Более подвержены риску заболевания лица мужского пола в возрасте 30 – 40 лет.Оссифицирующий миозит развивается постепенно на фоне фибромиозита. Соединительная ткань, которая замещает поврежденные мышечные волокна, постепенно трансформируется в неоднородную массу и пропитывается различными минералами и веществами. Когда накапливаются большие количества солей фосфорной кислоты, калия, кальция, начинается процесс окостенения. Окостеневшие участки мышц часто сращиваются с близлежащими костями, деформируя скелет.
Основными признаками полифибромиозита оссифицирующего миозита являются:
- уплотнение участков мышцы;
- деформация конечностей;
- снижение подвижности;
- появление сильных болей, особенно при движении.
Симптомы миозита
 Симптомами, которые указывают на миозит, являются:
Симптомами, которые указывают на миозит, являются:
- общие признаки травмы, инфекции;
- слабость и быстрая утомляемость;
- боль;
- снижение подвижности;
- изменение консистенции мышц;
- кожные изменения;
- изменения чувствительности;
- появление контрактур и аномальных положений конечностей.
В первые дни появляются:
- гиперемия (покраснение ) кожи;
- отек;
- болезненность;
- подкожные кровоизлияния;
- гематомы;
- иногда повышается местная температура.
Когда развивается воспалительный процесс в мышце, первым страдает тонус мышц. Мышечные волокна теряют способность быстро и в полную силу сокращаться и расслабляться. Больной ощущает нарастающую слабость в пораженной части тела. При миозите конечностей сложно поднимать руки выше головы или передвигать ноги. Слабость может достигнуть такой степени, что больному становиться сложно вставать со стула или кровати.
Основной характеристикой миозита является боль в пораженной мышце или группе мышц. Воспалительный процесс приводит к разрушению мышечных волокон и скоплению большого количества активных веществ в очаге воспаления, которые раздражают нервные окончания. Боль варьирует от умеренной до сильной в зависимости от места поражения и стадии заболевания.
При шейном миозите появляется острая боль при поворотах головы, при жевании. Иногда она распространяется к затылку и вискам или вниз в межлопаточную область.
При грудном миозите боль возникает при движениях грудной клетки (при глубоких вдохах и выдохах ) и при поворотах.
Миозит поясничной области вызывает умеренные боли, ноющего характера. Зачастую его путают с радикулитом . Но боль при радикулите более интенсивна.
Миозит конечностей вызывает усиление боли при ходьбе, при поднятии предметов. Часто больные стараются держать пораженную конечность в положении, которое приносит меньше боли.
Любая боль усиливается при движениях, при неудобных позах, при пальпации, при новых травмах, при воздействии низких температур, при смене погодных условий.
При хроническом миозите в период ремиссии боль утихает и даже может исчезнуть.
На подвижность пораженной области влияют несколько факторов. Во-первых, сильные боли сковывают движения, амплитуда их снижается. Во-вторых, разрушение большого количества мышечных волокон и замещение их соединительной тканью снижает эластичность мышц, соответственно снижается и сократительная способность. Движения становятся замедленными и неполными. Также движения лимитируются, когда начинается окостенение поврежденного участка мышцы. Если оссифицированные (окостеневшие ) участки срастаются с костями, движения сводятся к минимуму.
При полимиозите, могут поражаться и жизненно важные группы мышц (диафрагма, мышцы глотки ). При этом больному становится тяжело глотать, говорить и дышать.
В зависимости от стадии процесса, консистенция мышц различна. При воспалении, когда разрушаются мышечные волокна и накапливаются различные вещества в межклеточном пространстве, мышца становится плотной и немного увеличенной в объеме. Когда происходит реабсорбция (обратное впитывание ) всех этих веществ, мышца становится дряхлой, мягкой. При замещении мышечной структуры соединительной тканью пальпаторно обнаруживаются немного уплотненные узелки, которые могут увеличиваться в размерах. При оссифицирующем миозите пальпация выявляет твердые структуры, которые находятся в толще мышц или соединены с костью. При любом виде миозита пальпация вызывает болевые ощущения.
Часто миозит сопровождается кожными изменениями, и тогда он носит название дерматомиозита. Воспалительный процесс вовлекает все близлежащие ткани, особенно кожу. На коже появляются различные высыпания, красноватого и фиолетового оттенка. Они немного возвышаются над поверхностью кожи, придавая ей бугристый вид.
При вовлечении в воспалительный процесс внутримышечных нервных волокон и дистальных окончаний нервов изменяется чувствительность. Иногда наблюдается гиперчувствительность на любые внешние раздражители.
Нарушение структуры мышечной ткани, рубцевание и окостенение приводит к укорочению мышц, изменению формы и образованию различных контрактур. Из-за этого появляются различные искривления и анормальные положения тела. При шейном миозите появляется тортиколлис (искривление шеи ), при грудном миозите – сколиоз .
Диагностика миозита
 Лечение миозита находится в компетенции таких врачей как невропатолог, ревматолог и терапевт. Первоначально, при появившихся болях в спине, шее или ногах, необходимо обратиться к терапевту. Далее, в зависимости от этиологии заболевания, семейный врач рекомендует консультацию того или иного специалиста. Так, при миозите вследствие аутоиммунных заболеваний рекомендуется обратиться к ревматологу; при миозите во время простудных заболеваний – к терапевту; при нейро– и дерматомиозите – к невропатологу.
Лечение миозита находится в компетенции таких врачей как невропатолог, ревматолог и терапевт. Первоначально, при появившихся болях в спине, шее или ногах, необходимо обратиться к терапевту. Далее, в зависимости от этиологии заболевания, семейный врач рекомендует консультацию того или иного специалиста. Так, при миозите вследствие аутоиммунных заболеваний рекомендуется обратиться к ревматологу; при миозите во время простудных заболеваний – к терапевту; при нейро– и дерматомиозите – к невропатологу.Диагностика миозита кроме опроса и осмотра, может включать различные лабораторные и инструментальные обследования, поэтому пациент должен заранее быть готов к значительным временным и материальным затратам.
Диагностика миозита включает:
- опрос;
- осмотр;
- лабораторные исследования (ревмопробы );
- инструментальные исследования;
- биопсию.
Опрос
Включает данные о том, как началась болезнь и что ей предшествовало.Врач может задать следующие вопросы:
- «Что беспокоит на данный момент?»
- «Что было первым симптомом?»
- «Имела ли место температура?»
- «Предшествовало ли болезни переохлаждение, травмы?»
- «Какими заболеваниями еще страдает пациент?»
- «Чем болел пациент месяц или пару месяцев назад?»
- «Чем болел в детстве?» (например, болел ли в детстве ревматической лихорадкой? )
- «Есть ли какие-то наследственные патологии в семье?»
Осмотр
Изначально врач визуально осматривает место боли. Его внимание привлекает покраснение кожных покровов над мышцей или наоборот их побледнение. При дерматомиозите на коже в области разгибательных поверхностей (суставов ) образуются красные, шелушащиеся узелки и бляшки. Внимание врача могут привлечь ногти, так как одним из ранних признаков дерматомиозита является изменение ногтевого ложа (покраснение и разрастание кожи ). Длительно текущие миозиты сопровождаются атрофией мышцы. Над атрофированной мышцей кожа бледная со скудной сетью кровеносных сосудов.Далее врач приступает к пальпации (ощупыванию ) пораженной мышцы. Это делается с целью оценить тонус мышцы и выявить болезненные точки. В остром периоде болезни мышца напряжена, так как развивается ее гипертонус. Гипертонус является своеобразной защитной реакцией скелетной мускулатуры, поэтому при простудах и стрессах мышца всегда напряжена. Например, при шейном миозите мышцы бывают настолько напряжены, что затрудняют движение пациента. Иногда могут даже нарушаться процессы глотания, если воспалительный процесс охватил большую часть мышц шеи.
Болезненность мышцы может быть как общей, так и локальной. Например, при инфекционном гнойном миозите выявляются локальные болезненные точки, которые соответствуют гнойным очагам. При полифибромиозите болезненность усиливается по направлению к суставу, то есть в местах прикрепления мышцы.
При полимиозите болевой синдром умеренно выражен, зато прогрессирует мышечная слабость. В клинической картине оссифицирующего миозита боль умеренная, зато мышцы очень плотны, а во время их ощупывания выявляются плотные участки. Выраженный болевой синдром наблюдается при нейромиозите, когда вместе с мышечной тканью поражаются и нервные волокна.
Ревмопробы
Ревмопробами называются те анализы, которые направлены на выявление системных или локальных ревматических заболеваний.Такими заболеваниями могут быть:
- ревматоидный артрит;
- системная красная волчанка;
- полимиозит;
- полифибромиозит;
- миозит с включениями и другие.
В диагностике миозита ревмопробы включают определение следующих показателей:
- С-реактивного белка;
- антистрептолизина-О;
- ревмофактора;
- антинуклеарных антител (АНА );
- миозит-специфических аутоантител.
Повышенная концентрация С-реактивного белка наблюдается при различных воспалительных процессах в организме. С-реактивный белок является маркером острой фазы воспаления, поэтому определяется при острых инфекционных миозитах или при обострениях хронических. Определяя уровень этого белка можно оценить эффективность принятого лечения. Однако в целом С-реактивный белок является лишь показателем инфекционного процесса и не играет важную роль в дифференциальной диагностике миозита.
Антистрептолизин-О
Представляет собой антитело (белок
), которое вырабатывается в ответ на присутствие в организме стрептококка , а точнее на продуцируемый им фермент – стрептолизин (отсюда и название
). Является важным диагностическим критерием при ревматизме и ревматоидном артрите. Таким образом, повышенный титр этих антител говорит в пользу ревматического миозита.
Ревмофактор
Ревмофактор – это антитела, которые вырабатываются организмом к собственным белкам (иммуноглобулинам
). Повышенные значения ревмофактора наблюдаются при аутоиммунных патологиях, дерматомиозите, ревматоидном серопозитивном артрите. Однако существуют случаи, когда ревмофактор отрицателен. Это наблюдается при серонегативном ревматоидном артрите или у детей с ювенильным артритом. Важное диагностическое значение имеет количественное определение ревмофактора до и после лечения.
Антинуклеарные антитела
Семейство аутоантител, которое врабатывается к компонентам собственных белков, а именно к ядрам клеток. Наблюдаются при дерматомиозите, склеродермии и при других системных коллагенозах.
Миозит-специфические аутоантитела
Миозит-специфические аутоантитела (
MSA) являются маркерами таких идиопатических миозитов, как:
- дерматомиозит;
- полимиозит;
- миозит с включениями.
Самые распространенные антитела - это:
- Анти Jo-1 – выявляется у 90 процентов людей, страдающих миозитом;
- Анти-Mi-2 – наблюдается у 95 процентов людей с дерматомиозитом;
- Анти-SRP – выявляется у 4 процентов людей с миозитом.
Биопсия и морфологическое исследование мышечной ткани
Биопсия – метод диагностики, при котором происходит прижизненный забор кусочков тканей (биоптата ), с последующим их изучением. Целью биопсии в диагностике миозитов является определение структурных изменений в мышечной ткани, а также в окружающих ее сосудах и соединительной ткани.Показаниями к биопсии являются:
- инфекционные миозиты;
- полимиозиты (и как их разновидность дерматомиозиты );
- полифибромиозиты.
Мази для лечения негнойного инфекционного миозита
| Представители | Механизм действия | Как назначается |
| фастум гель (активное вещество кетопрофен ). Синонимы – быструм гель. | оказывает противовоспалительное действие, а также обладает высокой анальгетической активностью | наносят на кожу над очагом воспаления небольшое количество геля (5 см ) и втирают два – три раза в сутки |
| апизартрон (мазь не назначается в острых периодах ревматических болезней ) | входящий в состав препарата экстракт горчичного масла вызывает прогревание тканей, улучшает местный кровоток и расслабляет мышцы, также обладает противовоспалительным эффектом | полоску мази в 3 – 5 см наносят на воспаленный участок и медленно втирают в кожу |
| долобене - комбинированный препарат, который содержит диметилсульфоксид, гепарин и декспантенол. | кроме противовоспалительного и анальгезирующего действия, оказывает антиэкссудативный эффект, то есть устраняет отек | столбик геля длиной в 3 см наносят на очаг воспаления и втирают легким движением. Процедуру повторяют 3 – 4 раза в день |
При обширных миозитах, которые затрагивают несколько групп мышц и которые сопровождаются повышенной температурой и другими симптомами простуды лечение назначается в инъекционной форме (уколы ).
Уколы для лечения негнойного инфекционного миозита
| Представители | Механизм действия | Как назначается |
| диклофенак | оказывает противовоспалительный и обезболивающий эффект | по одному уколу (3 мл ) внутримышечно через день в течение 5 дней. |
| мелоксикам | благодаря селективному ингибированию образования медиаторов воспаления оказывает выраженный противовоспалительный эффект с минимальным развитием побочных эффектов | по одной ампуле (в 15 мг ) в сутки, внутримышечно в течение 5 дней, потом переходят на таблетированную форму препарата |
| мидокалм | оказывает миорелаксирующее (расслабляет напряженные мышцы ) действие | вводится внутримышечно по одной ампуле (100 мг вещества ) два раза в день. Таким образом, суточная доза составляет 200 мг |
Таблетки для лечения негнойного инфекционного миозита
| Представители | Механизм действия | Как назначается |
| апонил (действующее вещество – нимесулид ) | как и все нестероидные противовоспалительные оказывает противовоспалительный и анальгезирующий эффект, а также обладает и жаропонижающим действием | суточная доза препарата составляет 200 мг, что равняется 2 таблеткам по 100 мг, или 4 – по 50 мг. Дозу разделяют на 2 – 4 приема, запивая таблетку небольшим количеством воды |
| траумель С (препарат растительного происхождения ) | оказывает анальгезирующее и противоэкссудативное действие | по одной таблетке трижды в день. Таблетка ставится под язык, до полного рассасывания |
Чаще же всего лечение миозита комбинированное, то есть медикаменты назначаются как местно (в виде мази ), так и системно (в виде таблеток или уколов ).
Лечение полимиозита и его форм (дерматомиозита)
Основными препаратами в лечении полимиозита и его формы дерматомиозита являются глюкокортикостероиды. Препаратом выбора является преднизолон, который в остром периоде болезни назначается в виде уколов.Уколы для лечения полимиозита и его формы дерматомиозита
При неэффективности проводимой терапии проводится так называемая puls-терапия, которая заключается во введении сверхвысоких доз глюкокортикоидов (1 – 2 грамма
) внутривенно на короткий период (3 – 5 дней
). Данная терапия проводится исключительно в стационаре.
Преднизолон в таблетках назначается в качестве поддерживающей терапии, после достижения ремиссии. Также в таблетированной форме назначаются метотрексат и азатиоприн. Эти препараты относятся к группе иммунодепрессантов и назначаются в самых тяжелых случаях и при неэффективности преднизолона.
Таблетки для лечения полимиозита и его формы дерматомиозита
| Представители | Механизм действия | Как назначается |
| преднизолон | оказывает противовоспалительное, противоаллергическое и иммунодепрессивное действие | в период поддерживающей терапии 10 – 20 мг в сутки, что равняется 2 – 4 таблеткам по 5 мг. Эта суточная доза разбивается в два приема и принимается в первой половине дня. |
| метотрексат | цитостатический препарат, который оказывает иммуносупрессивное действие | назначается по 15 мг внутрь в сутки, постепенно увеличивая дозу до 20 мг. После достижения дозы в 20 мг переходят на инъекционные формы метотрексата. |
| азатиоприн | также оказывает иммуносупрессивное действие | назначается внутрь, начиная с 2 мг на кг веса в сутки. Лечение проводится под ежемесячным контролем анализа крови. |
Поскольку при полиомиозите наблюдается диффузное воспаление мышц, то назначение мазей нецелесообразно.
Лечение оссифицирующего миозита
При оссифицирующем миозите консервативное лечение эффективно лишь в начале болезни, когда еще возможно рассасывание кальцината. В основном лечение этого вида миозита сводится к хирургическому вмешательству.Массаж и втирание мазей противопоказаны.
Лечение полифибромиозита
 Лечение полифибромиозита включает противовоспалительные средства, уколы лидазы, массаж и физиопроцедуры.
Лечение полифибромиозита включает противовоспалительные средства, уколы лидазы, массаж и физиопроцедуры.Мази для лечения полифибромиозита
Уколы для лечения полифибромиозита
В форме таблеток назначаются противовоспалительные средства, которые целесообразны только в острой фазе болезни.
Таблетки для лечения полифибромиозита
| Представители | Механизм действия | Как назначается |
| бутадион | оказывает выраженный обезболивающий и противовоспалительный эффект. | по 150 – 300 мг (это одна – две таблетки ) 3 – 4 раза в день через 30 минут после еды. |
| ибупрофен | оказывает выраженное противовоспалительное и анальгезирующее действие. | по 800 мг (это две таблетки по 400 мг или одна по 800 ) от двух до четырех раз в сутки. При этом суточная доза не должна превышать 2400 мг, то есть 6 таблеток по 400 мг, или 3 по 800. |
Лечение гнойного инфекционного миозита
Включает применение антибиотиков , болеутоляющих и жаропонижающих средств. В некоторых случаях показано хирургическое вмешательство.Мази с последующим их растиранием над пораженной поверхностью противопоказаны, так как могут способствовать распространению гнойного процесса на здоровые ткани.
Уколы для лечения гнойного инфекционного миозита
| Представители | Механизм действия | Как назначается |
| пенициллин | оказывает бактерицидное действие за счет угнетения синтеза клеточной стенки микроорганизмов. Активен как в отношении грамположительных, так и в отношении грамотрицательных бактерий | внутримышечно по 300.000 ЕД. 4 раза в сутки (через каждые 6 часов ) |
| тетрациклин | внутримышечно по 200.000 ЕД. 3 раза в сутки (через каждые 8 часов ) | |
| цефазолин | обладает широким спектром антимикробного действия | внутримышечно по 1 грамму 4 раза в сутки (через каждые 6 часов ) |
Таблетки для лечения гнойного инфекционного миозита
Лечение миозита при аутоиммунных заболеваниях
Параллельно с лечением основного заболевания, которому сопутствует миозит (системная красная волчанка, склеродермия ) проводится симптоматическая терапия миозита. Она заключается в приеме обезболивающих и противовоспалительных средств, в острой фазе соблюдается пастельный режим.Мази для лечения миозита при аутоиммунных заболеваниях
| Представители | Механизм действия | Как назначается |
| найз гель | входящий в состав мази нимесулид оказывает анальгезирующее и обезболивающее действие | не втирая гель наносят тонким слоем на область болезненности. Процедуру повторяют от 2 до 4 раз в сутки |
| вольтарен мазь и гель (активное вещество диклофенак ) | оказывает выраженное противовоспалительное действие, также устраняет боль | 1 г мази (горошина величиной с лесной орех ) наносят над очагом воспаления, втирают в кожу 2 – 3 раза в сутки. Разовая доза - 2 грамма. |
| финалгель | 1 г геля наносят на кожу над пораженной областью и слегка втирают. Процедуру повторяют 3 – 4 раза в день. |
Уколы для лечения миозита при аутоиммунных заболеваниях
| Представители | Механизм действия | Как назначается |
| амбене | комбинированный препарат, который кроме противовоспалительного действия производит противоревматический эффект. | по одной инъекции (одна инъекция включает 2 мл раствора А и 1 мл раствора В ) внутримышечно через день. Курс лечения – 3 укола, после чего делают перерыв в 3 – 4 недели, а затем курс можно повторить. |
| баралгин М | кроме анальгезирующего и противовоспалительного действия производит спазмолитический (расслабляющий ) эффект. | внутримышечно вводят по одному уколу (5 мл ) от одного до двух раз в день. Максимальная суточная доза – 10 мл (2 укола ). |
Таблетки для лечения миозита при аутоиммунных заболеваниях
| Представители | Механизм действия | Как назначается |
| кетопрофен | производит обезболивающий и противовоспалительный эффект | в остром периоде болезни назначается доза в 300 мг в сутки, что равняется 3 таблеткам по 100 мг. В период поддерживающей терапии назначается по 150 – 200 мг в сутки. |
| нурофен | оказывает мощный анальгезирующий эффект | назначается по 400 – 800 мг от 3 до 4 раз в сутки. |
| флугалин | обладает противовоспалительным и болеутоляющим действием. | внутрь по одной таблетке 2 – 4 раза в день после еды с небольшим количеством еды. Курс лечения 2 – 3 недели. |
Лечение миозита народными средствами
 Терапия миозита народными средствами заключается в применении мазей, масел, растворов и настоек на спирту для растирания. Широко используются противовоспалительные компрессы и изоляция теплом пораженного участка мышц. Проведение данных манипуляций требует ограничения двигательной активности и максимальное обеспечение покоя. С болевым синдромом при миозите помогают справиться травяные настои, перед употреблением которых следует проконсультироваться с лечащим врачом.
Терапия миозита народными средствами заключается в применении мазей, масел, растворов и настоек на спирту для растирания. Широко используются противовоспалительные компрессы и изоляция теплом пораженного участка мышц. Проведение данных манипуляций требует ограничения двигательной активности и максимальное обеспечение покоя. С болевым синдромом при миозите помогают справиться травяные настои, перед употреблением которых следует проконсультироваться с лечащим врачом.Для исключения возникновения аллергических реакций при наружном применение народных средств перед лечением следует провести тест. Тестирование заключается в нанесение приготовленного состава на небольшой участок кожи. В случае возникновения покраснений, волдырей или сыпи стоит отказаться от применения выбранного рецепта.
Компрессы
Для снятия мышечной боли в народной медицине используется:- компресс из капусты;
- компресс из отварного картофеля;
- компрессы с использованием таких растений как ромашка, донник, липа, хвощ.
Для этой процедуры вам потребуются: 2 столовые ложки питьевой соды, 2 листа капусты белокочанной, детское мыло. Капусту следует обдать горячей водой, в которой предварительно была растворена 1 ложка соды. Далее нужно намылить листья мылом, посыпать оставшимся количеством соды и приложить к месту, которое вас беспокоит. Для усиления эффекта на область больной мышцы следует наложить согревающую повязку. Время действия компресса – 30 – 40 минут.
Компресс из отварного картофеля
Еще одним рецептом при миозите является компресс из отварного картофеля, для которого вам понадобятся: 3 – 5 отваренных в кожуре картофелин, одеколон, теплый шарф, чистая ткань. Разомните картошку и приложите через 2 слоя ткани к больному месту, после чего замотайте картофельный компресс шарфом. Действие компресса можно продлить, постепенно снимая тканевые слои. После того как картофель остыл, массу следует удалить, а приносящую дискомфорт область растереть, используя одеколон. Данную процедуру предпочтительнее всего проводить на ночь, для того чтобы дать разогретым мышцам отдых.
Компрессы из трав
Положительный эффект оказывают компрессы с использованием таких растений как ромашка, донника, липа, хвощ. Сухие растения следует положить в марлевый мешочек, запарить кипятком и обеспечить достаточное количество тепла, накрыв полиэтиленом и хорошо укутав больную область. Соблюдение всех рекомендаций при нанесении компрессов по рецептам народной медицины позволяет достичь положительного эффекта и значительно снизить мышечные боли.
Мази
Втирание мазей, приготовленных в домашних условиях, оказывает положительное воздействие, уменьшая болевые ощущения. Также мази используются в качестве основного ингредиента в компрессах, которые следует делать на ночь, обеспечив хорошую термоизоляцию.Женьшеневая мазь
Для того чтобы приготовить женьшеневую мазь, вам понадобятся: 20 грамм поваренной соли, 20 грамм высушенного корня женьшеня, 100 грамм медвежьего жира (продается в аптеке
), который можно заменить гусиным или свиным салом. Корень женьшеня следует измельчить и смешать с растопленным на водяной бане жиром и солью. Полученным составом нужно натирать больные места, применяя спиралевидные или прямолинейные движения снизу вверх.
Мазь на основе полевого хвоща и нутряного свиного сала
Следует взять 20 грамм сушеной травы и 80 грамм жировой основы и растереть массу в стеклянной или пластиковой посуде. Полученное средство втирать в те области, которые вас беспокоят. Также в качестве ингредиента для изготовления мазей на основе свиного сала или сливочного масла можно использовать такие растения как лаванда, листья эвкалипта, мята перечная, шалфей, чистотел.
Настойки
В качестве средства для растирания при лечении миозита используются изготовленные на спирту настойки с добавлением различных растительных компонентов. Настойки оказывают противовоспалительное, антибактериальное и болеутоляющее действие.Настойка на луке и камфорном масле
Для того чтобы приготовить это средство, нужно взять 2 крупные луковицы, 125 миллилитров (полстакана
) 70 процентного медицинского спирта и 1 литр камфорного масла. Лук следует измельчить и соединить со спиртом. Спустя два часа добавить в получившуюся массу масло и поставить настаиваться на десять дней, исключив доступ света. Состав может быть использован в качестве средства для растираний и компрессов.
Настойка из цветков сирени
Вам понадобятся 100 грамм свежей сирени и 500 миллилитров (два стакана
) 70 процентного медицинского спирта. Цветки заливаются спиртом и хранятся в течение недели в темном месте. Использовать для компрессов и растираний раз в сутки. Также в качестве ингредиентов для приготовления настоек может быть использована сухая или свежая ромашка, порошок бодяги. Одним из преимуществ настоек является их длительный срок хранения.
Масла
Изготовленные по рецептам народной медицины масла используются для массажей и растираний при возникновении обострений при миозите. Масла оказывают расслабляющее и согревающее воздействие на мышцы, способствуя снижению уровня болевых ощущений.Масло перцовое
Для того чтобы его приготовить, следует взять два небольших стручка жгучего перца и 200 миллилитров растительного масла. Перец нужно измельчить при помощи ножа или мясорубки вместе с семенами и залить маслом. Перелить состав в посуду из стекла и хранить в темном месте на протяжении 7 - 10 дней. По мере возникновения болей, нужно втирать перцовое масло в больные места, соблюдая меры предосторожности, так как, попав на слизистую, состав может вызвать сильное жжение.
Травяное масло
Для приготовления травяного масла понадобится:
- 700 миллилитров (три стакана ) нерафинированного растительного масла;
- 2 столовые ложки березового гриба;
- по одной столовой ложке таких растений как корень аира, трава адониса, бессмертник, зверобой, мелисса, тысячелистник, подорожник, череда, рябина обыкновенная, овес посевной, чистотел.
Отвары
При лечении миозита отвары, приготовленные на основе лекарственных трав, принимаются внутрь в соответствии с данными в рецепте указаниями. Основной эффект отваров заключается в их успокоительном действии на организм. Также травяные настои помогают уменьшить воспалительные процессы и снизить болевые ощущения.Отвар из плодов физалиса
Для его приготовления вам потребуется: 20 штук свежих или 20 грамм сухих плодов физалиса, 500 миллилитров дистиллированной воды. Плоды заливаются жидкостью и доводятся до кипения. После чего следует продолжать кипячение на слабом огне в течение 15 - 20 минут. Далее следует снять отвар, процедить, охладить и принимать по четверти стакана, 4 - 5 раз в день, перед приемами пищи. Через месяц следует сделать перерыв на 10 дней, после чего продолжить лечение.
Отвар из ивовой коры
Для того чтобы приготовить данное средство, следует взять 1 столовую ложку коры ивы и залить стаканом воды. Далее поместить состав на водяную баню и довести до кипения. Получившееся количество отвара следует разделить на 5 частей, которые употребить в течение дня. Продолжать курс нужно в течение 40 дней, после чего следует сделать перерыв на две недели.
Профилактика миозита
Что нужно делать?
Для профилактики миозита необходимо:- соблюдать сбалансированный рацион питания ;
- соблюдать водный режим;
- вести активный образ жизни, но в то же время избегать чрезмерных физических нагрузок;
- своевременно лечить простуды и другие инфекционные заболевания (нельзя переносить болезни на ногах и допускать их осложнений ).
Препятствовать воспалительному процессу в мышцах помогают жирные полиненасыщенные кислоты.
Достаточное количество полиненасыщенных кислот содержится в:
- лососевых видах рыб (семге, горбуше, кете );
- сельди;
- палтусе;
- тунце.
К таким продуктам относятся:
- морковь;
- свекла;
- картофель.
Водный режим
Питьевой режим очень важен при профилактике миозитов. Количество выпиваемой за день жидкости не должно быть меньше двух литров. Кроме некрепкого зеленого чая следует разнообразить питье морсами и компотами. Уменьшить отек в тканях помогает отвар шиповника.
Физическая активность
Для профилактики миозита следует придерживаться следующих пунктов:
- проводить больше времени на свежем воздухе;
- чередовать физические нагрузки с отдыхом;
- закалять организм;
- следить за осанкой;
- при длительной работе за компьютером делайте каждый час гимнастику для мышц спины и шеи.
Чего нужно избегать?
Для профилактики миозита следует исключить:- сидячий образ жизни;
- длительные нагрузки на одну группу мышц;
- пребывание на сквозняках;
- переохлаждение организма.
Определения
Мононейропатия . Изолированное поражение периферических нервов, например, при сдавлении, травме, нарушении кровоснабжения (поражение vasa vasorum ).
Системные заболевания, поражающие нервы, чувствительные к сдавлению, например сахарный диабет, или патологические состояния, вызывающие диффузные нарушения сосудистого русла (васкулиты), способны вызывать мультифокальную нейропатию (или множественную полинейронатию ).
Полинейропатия . Одновременное множественные поражение периферических нервов вследствие воспалительных процессов, метаболических нарушений или токсических воздействий. Клинически проявляется диффузным, симметричным поражением периферических нервов. В первую очередь страдают дистальные отделы конечностей, причем нижние конечности поражаются раньше верхних.
Мононейропатии
Наиболее часто встречаются следующие мононейропатии.
Синдром запястного канала
Компрессия срединного нерва в области запястья при его прохождении через канал может произойти:
- изолированно; например, у пациентов с избыточными физическим нагрузками (связанными с характером трудовой деятельности)
- при заболеваниях, характеризующихся повышенной чувствительностью нервных стволов к внешнему воздействию (сдавлению)
- при сдавлении нервного ствола в области запястного канала гипертрофированными тканями (табл. 1).
Таблица 1. Состояния, ассоциированные с синдромом запястного канала
Клинические проявления синдрома запястного капала:
- боль в кисти или предплечье, особенно ночью или при напряжении
- парез (паралич) и гипотрофия мышц возвышения большого пальца (thenar)
- снижение чувствительности в зоне иннервации срединного нерва (рис. 1)
- парестезии по ходу срединного нерва, которые возникают при постукивании в области запястного канала (симптом Тинеля )
- как правило, двустороннее поражение.
Рис. 1. Распределение зон иннервации срединного, локтевого и лучевого нервов на поверхности плеча и предплечья
Диагноз может быть подтвержден при помощи электрофизиологических исследований. Определение содержания в крови глюкозы, гормонов щитовидной железы, СОЭ, могут помочь в установлении правильного диагноза.
Лечение определяется тяжестью состояния больного. Основные лечебные мероприятия:
- фиксация мышц, особенно ночью, в частично вытянутом состоянии, кисть должна при этом находиться в состоянии разгибания
- мочегонные средства — эффект неясен
- введение кортикостероидов в просвет запястного канала
- хирургическая декомпрессия срединного нерва.
Нейропатия локтевого нерва
Локтевой нерв может быть подвержен сдавлению на различном уровне, однако наиболее часто это случается в области локтевого сустава.
Клинические проявления:
- боли и/или парестезии (покалывающего характера), распространяющиеся от локтя вниз по локтевой поверхности к предплечью
- паралич или слабость внутренних мышц кисти (поражение мышц возвышения большого пальца)
- снижение чувствительности в зоне иннервации локтевого нерва (рис. 1)
- при хроническом поражении формируется когтистая кисть .
Определение скорости проведения импульса при помощи электронейрографического исследования позволяет точно установить локализацию поражения локтевого нерва.
При нетяжелом поражении может быть эффективна фиксация руки на ночь, выпрямленной в локтевом суставе, что обеспечивает уменьшение сдавления нервного ствола. При более тяжелом поражении положительный результат обеспечивает хирургическая декомпрессия или транспозиция локтевого нерва, однако полный регресс неврологической симптоматики наблюдается не всегда. Оперативное вмешательство показано при постоянной травматизации локтевого нерва, которая сопровождается постоянным болевым синдромом и/или прогрессирующими двигательными нарушениями (парез).
Парез лучевого нерва
Сдавление лучевого нерва в верхней части предплечья может привести к острому развитию синдрома «свисающей кисти» , при этом иногда наблюдается утрата чувствительности в зоне иннервации лучевого нерва (рис. 1). Как правило, данное поражение является следствием длительного пребывания предплечья в непривычном положении, например при свисающей в неудобном положении руке с поручня кресла при алкогольном опьянении («паралич субботнего вечера» ).
Парез плечевого сплетения
Помимо острой травмы плечевого сплетения (например, в результате родовой травмы или дорожно-транспортного происшествия у мотоциклистов) поражение плечевого сплетения может быть обусловлено другими причинами. Поражение верхнего отдела сплетения носит название паралича Эрба , а нижнего - паралича Клюмпке .
Добавочное ребро
Добавочное ребро или гипертрофированная соединительная ткань может быть причиной компрессии плечевого сплетения в области верхней апертуры грудной клетки. На определенном этапе развития неврологии и нейрохирургии имела место гипердиагностика данного состояния и, как следствие, высокая частота необоснованных хирургических вмешательств. На сегодняшний день считается, что оперативное вмешательство показано пациентам с нарастающим парезом внутренних мышц предплечья, выраженной утратой чувствительности (по ходу локтевого нерва) и с диагнозом, подтвержденным электрофизиологическими методами обследования. Визуализация плечевого сплетения при помощи МРТ обычно неэффективна. При рентгенографическом обследовании возможно выявление добавочного ребра, но сдавление нервного ствола фиброзной тканью визуализировать не удается.
Опухоль Пенкоста
Бронхогенная карцинома верхушки легкого может прорастать в нижние корешки плечевого сплетения, вызывая усиливающуюся боль в одноименной руке, дистальный паралич и гипотрофию, а также снижение чувствительности в дерматомах С7, С8 и Th10. Возможен также синдром Горнера вследствие поражения преганглионарных вегетативных волокон. Симптоматика имеет сходство с первичными и метастатическими опухолями.
Диагностические трудности возникают при поражении сплетения у больных с карциномой молочной железы после проведенного курса лучевой терапии, так как неврологический дефицит может быть следствием распространения опухоли или радиационной плексопатии .
Идиопатическая плечевая плексопатия (невралгическая амиотрофия или нейропатия плечевого нерва)
Состояние характеризуется острой болью в плече и предплечье. Очевидных причин этому нет, хотя заболевание может возникнуть после прививки или операции. После регресса болей (через несколько дней или недель) появляется частичный паралич и слабость окололопаточной группы мышц, а также более удаленных мышечных групп верхней конечности. Особенно подвержена поражению передняя лестничная мышца, атрофия которой сопровождается развитием крыловидных лопаток (рис. 2). Поражение, как правило, одностороннее, с минимальными чувствительными нарушениями. Электрофизиологические исследования зачастую малоэффективны, хотя могут выявляться признаки денервации пораженных мышц. Состав СМЖ не изменен. Специфического лечения не существует, у большинства больных через 1,5-2 года наступает спонтанное выздоровление.

Рис. 2.
Компрессия латерального кожного нерва бедра, проходящего под паховой связкой; характеризуется потерей чувствительности в соответствующей области (рис. 3). Начало заболевания связано, в частности, с изменением веса пациента (увеличение или уменьшение).
Рис. 3. Парестетическая мералгия. Схема распределения нарушений чувствительности при поражении латерального кожного бедренного нерва
Латеральный подколенный паралич
Подколенный нерв подвержен компрессионным повреждениям в области, где он огибает шейку малоберцовой кости. Проявляется синдромом свисающей стопы (вследствие пареза разгибателя стопы). Одновременно появляются слабость при тыльном разгибании и отведении стопы с утратой чувствительности разной степени выраженности. Данное состояние часто встречается у иммобилизованных пациентов и у пациентов с повышенной чувствительностью нервных стволов к сдавлению, например при сахарном диабете. Свисающая стопа может быть следствием поражения поясничного корешка (обычно L5). Следует отличать данный синдром от поражения малоберцового нерва, для которого характерна сохранная внутренняя ротация стопы, так как задняя большеберцовая мышца иннервируется большеберцовым нервом, а не малоберцовым. Однако требуется электрофизиологическое исследование для уточнения локализации поражения нерва. Повреждение малоберцового нерва обычно обратимо, так как вызывается нарушением проводимости (нейрапраксия ). Положительный эффект оказывает фиксация стопы лонгетой .
Мультифокальная нейропатия
Причины мультифокальной нейропатии (множественный мононеврит):
- злокачественная инфильтрация (карцинома или лимфома)
- васкулит или заболевание соединительной ткани:
- ревматоидный артрит
- системная красная волчанка
- узелковый периартрит
- гранулематоз Вегенера;
- саркоидоз
- диабет
- инфекционные заболевания:
- проказа
- опоясывающий герпес
- болезнь Лайма;
- наследственная нейропатия со склонностью к параличам от сдав-ления.
Наиболее частой причиной мультифокальной нейропатии является васкулит с болями, слабостью и гипестезией в зонах иннервации нескольких периферических нервов. Чаще поражаются нижние конечности. Поражения отдельных нервов постепенно накапливаются, проявляясь асимметричным поражение конечностей.
Полинейропатии
Диффузное поражение периферических нервов может быть разделено на группы с поражением двигательных, чувствительных или смешанных нервов. Существует патофизиологическая классификация полинейропатии, основным критерием которой является преобладание поражения миелиновой оболочки или непосредственно нервного ствола нерва (демнелинизирующая или аксональная нейропатия соответственно). Причины полинейропатии приведены в табл. 2.
Таблица 2. Причины полинейропатии
|
Наследственная предрасположенность Инфекционные заболевания Дифтерия Болезнь Лайма Синдром Гийена-Барре Хроническая воспалительная демиелинизирующая полинейропатия Саркоидоз Синдром Шегрена Васкулиты (например, волчанка, полиартериит) Новообразования Паранеопластические процессы Парапротеинемические процессы Метаболические расстройства Микседема Отложения амилоида Неправильное питание Дефицит витаминов, в особенности тиамина, ниацина и витамина В 12 Отравления Например, алкоголем, свинцом, мышьяком, золотом, ртутью, таллием, инсектицидами, гексаном Лекарственные препараты Например, изониазид, винкристин, цисплатин, метронидазол, нитрофураны, фенитоин, амиодарон |
У пациентов могут развиться онемение и/или парез дистальных отделов конечностей. Двигательные нарушения характеризуются вялыми парезами и мышечными атрофиями. Длительно развивающиеся нейропатии могут привести к деформации стоп и кистей рук (полая стопа — рис. 4 и когтистая кисть соответственно). Тяжелое поражение сенсорных волокон может сопровождаться развитием нейропатических язв и деформаций суставов (рис. 5). Возможны сопутствующие вегетативные расстройства. Клинические признаки такие же, как и при распространенном поражении периферических мотонейронов с вялыми параличами, гипотонией и снижением сухожильных рефлексов. Утрата проприоцептивной чувствительности в дистальных отделах конечностей может сопровождаться сенсорной атаксией . Характерно снижение болевой, температурной и тактильной чувствительности но типу «носков и перчаток». В ряде случаев можно обнаружить утолщение периферических нервов. Тактика обследования пациентов с полинейропатиями приведена в табл. 3.

Рис. 4.

Рис. 5. Нейропатия правой лодыжки (слева) и ступни (справа; артропатия Шарко)
Таблица 3. Обследование больного с нейропатией
|
Исследование крови Клинический анализ с подсчетом форменных элементов, СОЭ, содержание глюкозы, электролитов, мочевины, печеночных ферментов и гормонов щитовидной железы, витамина В 12 , электрофорез сывороточных белков, определение аутоантител Исследование мочи Микроскопический анализ для подтверждения васкулита, определение содержания глюкозы, порфиринов, белка Бена-Джонса Исследование СМЖ Повышенное содержание белка, в частности при воспалительных нейропатиях Нейрофизиологическое обследование Изучение скорости проведения по двигательным и чувствительным нервам и ЭМГ Рентгенография органов грудной клетки Для исключения саркоидоза, карциномы Специальные обследования для отдельных пациентов Биопсия периферических нервных волокон при неизвестном характере нейропатии и ухудшении состояния больного. Проводится для подтверждения наличия васкулита, проказы и хронической воспалительной демиелинизирующей полинейропатии. Биопсия костного мозга, обследование скелета при подозрении на миеломную болезнь. При определенных состояниях — специфические анализы крови, например при наследственных нейропатиях — анализ ДНК, при врожденных нарушениях метаболизма — обнаружение ферментов из лейкоцитов, при болезни Лайма — обнаружение антител к боррелии. |
Лечение больных с полинейропатиями определяется в первую очередь причинами заболевания. Пациенты с воспалительными полинейропатиями требуют госпитализации в специализированные отделения. Больному с острой воспалительной демиелинизирующей полинейропатией (синдром Гийена-Барре ) может потребоваться реанимационная помощь. Хроническая воспалительная демнелинизирующая полинейропатия (ХВДП) и полиневропатия при васкулитах требуют применения кортикостероидов и/или иммуномодулирующих препаратов, включая иммуносупрессоры (азатиоприн, циклофосфамид или циклоспорин), внутривенное введение иммуноглобулина или плазмаферез. Симптоматическое лечение позволяет снизить вероятность осложнений, включая нарушения вегетативных функций и болевые синдромы.
Важно отличать синдром Гийена-Барре и ХВПД — демиелини-зирующие заболевания периферических нервов от демиелинизации ЦНС (табл. 17.4).
Таблица 4. Заболевания, приводящие к демиелинизации. Классификация в соответствии с локализацией основного очага поражения и характером течения заболевания
Нейромышечный синапс
Миастения
Аутоиммунное заболевание, при котором у большинства пациентов выявляются циркулирующие антитела к ацетилхолиновым рецепторам нейромышечных синапсов (рис. 6). В качестве причины возможна патология тимуса (гиперплазия, атрофия или опухоль — тимома ). Данное заболевание встречается относительно редко, в среднем в год регистрируется 0,4 случая на 100 000, но так как большинство пациентов выживает, то количество заболевших достигает 1 на 10 000. Подвержены все возрастные группы.

Рис. 6.
Клинические проявления
- Диплопия с ограничением движения глазных яблок
- Слабость мимических мышц
- «Голос миастеника»
- Слабость при закрывании глаз
- Бульбарные нарушения:
- дисфагия (с попаданием пищи в носовые ходы)
- дизартрия (с носовым оттенком)
- Вовлечение дыхательных мышц (острые бульбарные и дыхательные расстройства, вызванные миастенией, требуют неотложной помощи)
- Слабость мышц шеи и конечностей, усиливающаяся к концу дня и после нагрузок («патологическая утомляемость» ).
Обследование
- Определение содержания антител к ацетилхолиновым рецепторам в сыворотке крови (у 15% пациентов результат анализа отрицательный).
- Проба с введением антихолинэстеразных препаратов: преходящее и быстро нарастающее улучшение состояния после внутривенного введения эдрофониума (антихолинэстеразный препарат короткого действия, блокирует катаболизм ацетилхолина, временно повышая его содержание). (В Российской Федерации используется тест с прозерином). Тест более эффективен при использовании метода двойного контрольного исследования. Ввиду возможных холиномиметических эффектов вследствие повышения уровня ацетилхолина следует обеспечить возможность экстренного введения атропина и проведения реанимационных мероприятий.
- ЭМГ, включая игольчатое исследование с отведением потенциала от отдельных волокон.
- Исследование функции щитовидной железы при сопутствующем тиреотоксикозе.
- У пациентов с тимомой выявляются антитела в тканях поперечнополосатых мышц.
- КТ переднего средостения для выявления гиперплазии тимуса.
Лечение
- Антихолинэстеразные препараты , например пиридостигмин, в качестве симптоматического лечения. Пациентам требуется постоянное увеличение дозировок препаратов, что может привести к развитию холиномиметических побочных эффектов с повышением слюноотделения, рвотой, болью в эпигастрии и диареей. В редких случаях возможно развитие холинергического криза
- Кортикостероиды (преднизолон) назначаются при заболевании средней тяжести, не поддающемуся другому лечению. Лечение начинается с низких дозировок с постепенным увеличением дозы, препарат применяют через день. В начале лечения возможно нарастание симптоматики. Стационарное лечение показано в начале применения кортикостероидов у пациентов с генерализованными формами заболевания. По мере наступления эффекта доза может быть снижена в соответствии с клинической картиной.
- Иммуносупрессоры (азатиоприн) используются в комбинации с кортикостероидами при заболевании средней тяжести.
- Тимэктомия показана при тимоме и у молодых пациентов на ранних стадиях заболевания для уменьшения потребности в лекарственной терапии и реже — для достижения полной ремиссии.
- Плазмаферез или внутривенное введение иммуноглобулина как средства подготовки к тимэктомии и при тяжелых формах заболевания.
Пациентам с миастенией следует избегать приема некоторых антибиотиков, таких как аминогликозиды, вследствие их блокирующего эффекта на уровне нейромышечного синапса.
Другие миастенические синдромы
Реже нейромышечный синапс может страдать в результате наследственного заболевания или вследствие наранеопластического процесса (миастенический синдром Ламберта-Итона).
Миопатии
Основные причины развития миопатий приведены в табл. 5. Клинически миопатия проявляется слабостью мышц туловища и проксимальных отделов конечностей. Возможны слабость мимических мышц и мускулатуры шеи, выявляемая при сгибании и/или разгибании. Походка становится неустойчивой. При приобретенном характере заболевания мышечная слабость может быть относительно умеренной, по крайней мере на ранних стадиях, а сухожильные рефлексы на протяжении длительного времени остаются сохранными.
Таблица 5. Причины возникновения миопатий
|
Наследственные факторы Мышечная дистрофия Метаболические миопатий Инфекционные заболевания Газовая гангрена Стафилококковый миозит Вирусная инфекция (вирусы гриппа, Коксаки, ЕСНО) Воспалительные процессы Полимиозит Дерматомиозит Саркоидоз Новообразования
Дерматомиозит - может быть следствием паранеопластического процесса Метаболические (приобретенные) расстройства Тиреотоксикоз Синдром Кушинга Остеомаляция Токсикоз (от приема лекарственных препаратов) Кортикостероиды Галотан - злокачественная гипертермия (редко) Другие лекарственные препараты |
Обследование больного с миопатией:
- анализ крови:
- СОЭ, аутоантитела (при приобретенных заболеваниях)
- креатинкиназа — уровень резко повышен вследствие высвобождения из поврежденных мышечных клеток
- биопсия мышц.
Клинические синдромы
Мышечные дистрофии
Дистрофинопатии
Заболевание обусловлено мутацией гена, связанного с Х-хромосомой и отвечающего за синтез мышечного белка дистрофина . Встречается у детей, подростков и у взрослых. Детская форма (мышечная дистрофия Дюшена ) протекает наиболее тяжело. У заболевших мальчиков уже в раннем детстве развивается слабость в проксимальных отделах конечностей. Характерным признаком является взбирание «лесенкой» (симптом Говерса ). Мышцы голеней могут казаться гипертрофированными (рис. 7) из-за замещения мышечных волокон соединительной тканью (псевдогипертрофия ). Дети обычно прикованы к креслу-каталке до подросткового возраста. Болезнь неуклонно прогрессирует, смерть наступает от сердечных или дыхательных осложнений в возрасте до 20 лет. Менее тяжелое течение наблюдается при дебюте заболевания в подростковом или взрослом возрасте (мышечная дистрофия Беккера ). Заболевание, как правило, не носит угрожающего для жизни характера, однако зачастую сопряжено с прогрессирующей инвалидизацией. В настоящее время имеется возможность диагностики миодистрофий при помощи молекулярного анализа гена дистрофина.

Рис. 7.
Другие мышечные дистрофии
Миотоническая дистрофия — заболевание с аутосомно-доминант-ным типом наследования, при котором у пациентов присутствует аномально длительное напряжение мышц (миотония). Проявляется невозможностью расслабления мышцы. Характерным признаком является перкуссионная миотония , которая выявляется постукиванием молоточком по мышце. Миотония может быть диагностирована при электромиографическом обследовании.
Типичные симптомы:
- двусторонний птоз
- слабость лицевых мышц
- паралич и слабость грудино-ключично-сосцевидных мышц
- ранняя катаракта
- сопутствующие эндокринные нарушения (сахарный диабет, облысение и атрофия яичек).
В качестве терапии при миотонии могут применяться фенитоин или мексилетин. При наследственной миотонии наблюдаются умеренно выраженные атрофия и слабость мышц.
Лице-лопаточно-плечевая мышечная дистрофия является аутосомно-доминантным заболеванием. У пациентов наблюдается двусторонняя слабость мимических мышц и крыловидное расположение лопаток. В дополнение к параличу и слабости проксимальных мышц верхних конечностей обычно имеются слабость мышц спины и тазового пояса, наблюдается неустойчивая походка и поясничный гиперлордоз. Реже при мышечных дистрофиях и врожденных миопатиях поражаются мышцы глазного яблока и глотки.
Другие наследственные заболевания
Метаболические нарушения, например гликогенозы , могут сопровождаться мышечной слабостью, нередко ассоциированной с миалгиями и спазмами.
При семейном периодическом параличе приступы выраженной мышечной слабости могут быть спровоцированы напряжением, употреблением пищи с высоким содержанием углеводов, длительным пребыванием на холоде. Заболевание может быть связано с гипо- и гиперкалиемией.
Приобретенные заболевания
Воспалительные миопатии
Полимиозит может развиваться как изолированно, гак и вместе с другими аутоиммунными поражениями соединительной ткани, например системным склерозом, фиброзирующим альвеолитом и синдромом ПТегрена.
Дерматомиозит является сопутствующим заболеванием при воспалительной миопатии с характерной фиолетовой (гелиотропной ) сыпью на лице. Ярко-красная сыпь может быть локализована в области суставов, передней поверхности грудной клетки, поверхностях разгибателей. У некоторых пациентов с дерматомиозитом, в частности у мужчин старше 45 лет, нередко имеется злокачественное новообразование, например карцинома бронхов или желудка. -
Клинические проявления воспалительной миопатии такие же, как при проксимальной миопатии, однако могут также присутствовать дисфагия как результат вовлечения мышц глотки, мышечные боли и гиперестезия. Возможны также артралгия и феномен Рейно.
После гистологического подтверждения диагноза проводится лечение кортикостероидами и иммуносупрессорами (например, азатиоприн). Пациентам требуется наблюдение в течение нескольких лет, у многих остается мышечная слабость. Один из гистологически диагностируемых вариантов заболевания — миозит с включением телец — лечению не поддается. Данное состояние — довольно частая форма приобретенных мышечных заболеваний, поражающая в основном пожилых людей. Характерной особенностью является избирательное поражение сгибателей пальцев кистей и четырехглавых мышц. Недостаточный эффект применения иммуносупрессоров послужил основой гипотезы о вторичности воспалительных реакций по отношению к дегенеративным изменениям мышечной ткани.
Неврология для врачей общей практики. Л. Гинсберг
Симптомом нервно-мышечного заболевания могут быть спазмы мышц или наоборот их резкое расслабление.
.jpg) Наследственные нервно-мышечные заболевания объединяют целую группу болезней, общей характеристикой которых является «записанные» в геноме нарушения в работе нервно-мышечного аппарата. Атрофия мышц, их чрезмерное сокращение или наоборот расслабление – все это может быть признаком болезней, передающихся по наследству.
Наследственные нервно-мышечные заболевания объединяют целую группу болезней, общей характеристикой которых является «записанные» в геноме нарушения в работе нервно-мышечного аппарата. Атрофия мышц, их чрезмерное сокращение или наоборот расслабление – все это может быть признаком болезней, передающихся по наследству.
Виды наследственных нервно-мышечных заболеваний
Наследственно нервно-мышечные заболевания включают в себя много разнообразных нарушений, которые разделяют на несколько групп:
- первичные прогрессирующие мышечные дистрофии или миопатии.
- вторичные прогрессирующие мышечные дистрофии.
- врожденные непрогрессирующие миопатии
- миотонии
- наследственные пароксизмальные миоплегии.
Первичные прогрессирующие мышечные дистрофии или миопатии
Миопатии включают в себя группу болезней, которые проявляются мышечной слабостью и дистрофией мышц, нарастающими с течением времени. При заболеваниях этой группы происходит в мышечных клетках, что приводит к атрофии мышечных волокон.
При миопатиях могут поражаться мышцы конечностей, таза, бедер, плеч, туловища, в зависимости от конкретной разновидности болезни. Наиболее распространены: юношеская форма Эрба-Рота, плече-лопаточно-лицевая форма Ландузи–Дежерина, псевдогипертрофическая форма Дюшенна.
При миопатиях мышечная сила и тонус мышц снижаются симметрично. Часто развивается псевдогипертрофия – увеличение мышц за счет разрастания жировой и соединительной ткани. Инфекции, интоксикации, стрессы могут ускорить течение болезни.
Первичные прогрессирующие мышечные дистрофии при активном течении могут приводить к инвалидизации и полному обездвиживанию.
Вторичные прогрессирующие мышечные дистрофии
При развитии этих заболеваний в первую очередь нарушается работа периферических нервов. Нарушается иннервация мышц, что приводит к возникновению мышечных дистрофий.
Вторичные прогрессирующие мышечные дистрофии включают три разновидности: врожденную, раннюю детскую и позднюю. Такая классификация основывается на времени появления первых признаков болезни. В зависимости от формы болезни она протекает более или менее агрессивно. В зависимости от этого люди, страдающие этой разновидностью генетических отклонений, доживают до 9-30 лет.
Непрогрессирующие миопатии
Миотония врожденная
Миотония врожденная (болезнь Томсена) - редкое наследственное заболевание, характеризующееся длительными тоническими спазмами мышц, возникающими вслед за начальными произвольными движениями.
Этиологи
К этой группе относятся болезни, также связанные с дистрофией мышц. Проблемы проявляются сразу при рождении. При этом обнаруживается «синдром вялого ребенка» - состояние, когда наблюдается мышечная вялость, двигательная заторможенность и отставание в двигательном развитии ребенка. Но непрогрессирующие миопатии отличаются от других видов наследственных нервно-мышечных заболеваний тем, что состояние с течением времени не ухудшается и болезнь не прогрессирует.
Миотонии
Для этой группы заболеваний характерны спазмы мышц в начале движения. В начале действия мышца сокращается и в течение 5-30 секунд не может расслабиться. После этого постепенное расслабление все-таки происходит и повторное движение делать уже немного легче. Но после отдыха все повторяется снова.
При этом заболевании спазм может задействовать мышцы лица, туловища, конечностей.
К наследственным миотониям относят дистрофическую миотонию, врожденную миотонию Томсена, атрофическую миотонию, парамиотонию и другие болезни.
Достаточно простым способом определить миотонию является симптом «кулака». При подозрении на миотонию врач просит быстро разжать кулак. Человек, страдающий этим генетическим заболеванием, не может сделать это быстро и без усилий. В качестве теста можно также предложить быстро разжать челюсти, встать со стула или открыть прищуренный глаз.
Люди, страдающие миотониями, часто имеют атлетическое сложение. Это связано с тем, что при этих заболеваниях определенные группы мышц гипертрофированы. Под воздействием холода и мышечный спазм обычно усиливается.
Как правило, человек, «имеющий» в своем геноме миотонию, может с ней сосуществовать. Таким людям просто стоит правильно выбрать профессию, в которой нет необходимости в резких движениях. Но есть разновидности миотонии, при которых существует риск развития инвалидности или внезапной смерти.
Миоплегии
Еще одной разновидностью наследственных нервно-мышечных заболеваний являются миоплегии. В этом случае характерной особенность болезни являются приступы мышечной слабости. Различают несколько форм пароксизмальных миоплегий: гипокалиемическую, гиперкалиемическую и нормокалиемическую.
При этом заболевании в мышечных клетках нарушается поляризация мембран и изменяются электролитические свойства мышц.
В момент приступа обычно возникает резкая слабость мышц рук ног или туловища. Иногда может появляться слабость глотки, гортани, дыхательных мышц, что может стать причиной летального исхода.
Все формы наследственных нервно-мышечных заболеваний плохо поддаются лечению. Но современная медицина продолжает искать способы воздействия на генетические заболевания. И в ближайшем будущем, возможно, будут разработаны эффективные способы воздействия на подобные генетические болезни.