Ліки від дистрофії м'язів. Прогресують м'язові дистрофії. Рідкісні форми Х-зчеплених міодистрофій
![]()
Опис:
М'язова дистрофія є групою хронічних спадкових захворювань скелетних м'язів людини, що виявляються слабкістю і дегенерацією м'язів. Існує дев'ять різних форм м'язової дистрофії. Вони різняться за такими характеристиками, як вік, у якому починається захворювання, локалізація уражених м'язів, вираженість м'язової слабкості, швидкість прогресування дистрофії та тип її наслідування. Найчастіше зустрічаються дві форми: м'язова дистрофія Дюшенна та міотонічна м'язова дистрофія.
Симптоми:
Дюшенна дистрофія. Х-хромосомна рецесивна мутація дистрофін-гену. Клінічні ознаки: початок віком до 5 років; прогресуюча слабкість м'язів тазового та плечового пояса; нездатність ходити після 12 років; кіфосколіоз; дихальна недостатність віком 20-30 років. Залучення інших систем органів: ; зниження інтелекту.
Беккер дистрофія. Х-хромосомна рецесивна мутація дистрофін-гену. Клінічні ознаки: початок у ранньому чи пізньому віці; повільно прогресуюча слабкість м'язів тазового та плечового пояса; збереження можливості ходити після 15 років; дихальна недостатність після 40 років. Залучення інших систем органів: кардіоміопатія.
Міотонічна дистрофія. Аутосомно-домінантний; розширення нестабільного ділянки ДНК хромосоми 19ql3,3. Клінічні ознаки: початок у будь-якому віці; повільно прогресуюча слабкість м'язів повік, обличчя, шиї, дистальних м'язів кінцівок; міотонія. Залучення інших систем органів: порушення серцевої провідності; психічні порушення; лобна; гонад.
Плече-лопатково-лицьова дистрофія.
Автосомно-домінантний; Часто мутації хромосоми 4q35. Клінічні ознаки: початок віком до 20 років; повільно прогресуюча м'язова слабкість лицьової області, плечового пояса, тильного згинання стопи. Залучення інших систем органів: гіпертензія; глухота.
Плечового та тазового пояса (можливі кілька захворювань). Аутосомно-рецесивний чи домінантний. Клінічні ознаки: початок із раннього дитинства до середнього віку; повільно прогресуюча слабкість м'язів плечового та тазового пояса. Залучення інших систем органів: кардіоміопатія.
Око-ковткова дистрофія. Автономно-домінантний (Французька Канада чи Іспанія). Клінічні ознаки: початок 50-60 років; повільно прогресуюча слабкість м'язів: зовнішніх очних, повік, обличчя та горлянки; крикофарингеальна ахалазія. Залучення інших систем органів: церебральні, очні.
Вроджена дистрофія. Включає кілька захворювань, у тому числі типи Фукуяма та церебро-окулярна дисплазія). Автосомно-рецесивний. Клінічні ознаки: початок при народженні; гіпотонія, затримка розвитку; в одних випадках - рання дихальна недостатність, в інших - більш сприятливий перебіг хвороби.
Причини виникнення:
Захворювання викликається аутосомно-домінантним геном з різкою експресивністю (можливість ризику передачі на родичів 1-го ступеня становить 50%). Захворювання викликається ампліфікацією, тобто підвищенням числа CTG - триплетів у певному локусі 19 хромосоми (тип 1 міотонічної дистрофії) або CCTG у 3 хромосомі (тип 2 міотонічної дистрофії). Тип 2 міотонічної дистрофії вивчений слабо. Вважається, що він зустрічається тільки в 2% випадків (але може бути значно частіше); не взаємопов'язаний із типом 1; найімовірніше не є причиною вроджених форм дистрофії, коли носієм є мати. Для 1 типу доведено, що кількість повторів нуклеотидів збільшується при передачі мутації з покоління до покоління. Тяжкість захворювання чітко корелює з чисельністю цих повторів. Найбільше їх кількість визначається при вродженій тяжкій формі захворювання. Виявлений механізм пояснює феномен антиципації - обтяження та все більш раннього початку хвороби у низхідних поколіннях. Наприклад, якщо генетичний аналіз показав, що має кілька повторів CTG, то в його дитини виявиться ще більше повторів цього триплету.
Лікування:
На сьогоднішній день не існує способів запобігти або сповільнити прогрес цього захворювання. Терапія спрямована головним чином на боротьбу з ускладненнями, такими як деформація хребта, що розвивається внаслідок слабкості м'язів спини, або схильність до пневмоній, обумовлена слабкістю дихальних м'язів. Фенітоїн, прокаїнамід, хінін застосовуються в лікуванні міотонії, але потрібна обережність у хворих із захворюваннями серця (небезпека погіршення серцевої провідності). Імплантація водія серцевого ритму необхідна хворим із синкопе або серцевою блокадою. При лікуванні серцевих порушень рекомендовано препарат Фенігідін. Застосування ортопедичних апаратів може зміцнити «висячі» стопи, стабілізувати гомілковостопні суглоби, зменшити частоту падінь. Добре підібране тренування також може мати позитивний вплив на перебіг цього захворювання. За наявності атрофії застосовують анаболічні стероїди (ретаболіл, неробол), загальнозміцнювальну терапію. У тих випадках, коли є значно виражена міотонічна симптоматика, призначають курси дифеніну по 0,03-0,05 г 3 рази на день тривалістю 2-3 тижні. Припускають, що дифенін має пригнічуючу дію на синаптичну провідність та знижує посттетанічну активність у м'язах. При підвищеній сонливості, яка нерідко супроводжує міотонічну дистрофію, позитивний ефект спостерігається при прийомі селегіліну. Рекомендовано також прийом деяких біологічно активних добавок: коферменту Q10 (100 мг/день), вітаміну Е (200 МО/день) та селену (200 мкг/день), лецитину (20 г/день).
Ефективне лікування від цього захворювання можливе лише за допомогою генної терапії, яка зараз інтенсивно розвивається. Численні експерименти показують поліпшення стану м'язових волокон під час лікування деяких форм м'язової дистрофії. При дистрофіях Дюшена та Беккера спостерігається недостатнє вироблення м'язового білка дистрофіну. Ген, відповідальний за виробництво цього білка, є найбільшим із усіх відомих генів, тому для проведення генної терапії вчені створили мініатюрну версію цього гена. Найкращими провідниками гена до м'язів вчені визнали аденовіруси. Тому всередину аденовірусу вони помістили потрібний ген і ввели його мишам, які страждають від нестачі дистрофіну. Результати досвіду виявилися обнадійливими. В інших подібних роботах носіями цього гена є ліпосоми, мікросфери, лактоферин. Оригінальний підхід до генотерапевтичного лікування МДД розробляється в Оксфордському університеті групою під керівництвом Кей Девіс (Kay Davies). Суть методу полягає у спробі дерепресії аутосомного гомолога дистрофіну – гена утрофіну, продукт експресії якого міг би бути здатний компенсувати нестачу дистрофіну у всіх групах м'язів. В ембріогенезі людини приблизно до семи тижнів розвитку дистрофін не експресується та його функцію у м'язах виконує білок утрофін. У проміжку між сьомою та 19 тижнями розвитку експресуються обидва білки і після 19 тижня відбувається заміщення м'язового утрофіну на дистрофін. Після 19 тижня ембріонального розвитку утрофін виявляється лише в ділянці нервово-м'язових контактів. Білок утрофін, маючи аутосомну локалізацію, нагадує дистрофін своїми N- і С-кінцевими доменами, що грають вирішальну роль у функції дистрофіну. Результати експериментів вказують на принципову можливість корекції дефектів у м'язових волокнах позбавлених дистрофіну за допомогою утрофіну. Встановлено, що два препарати (L-arginine та heregulin) збільшують виробництво білка атрофін у м'язових клітинах мишей. Підвищена кількість атрофіну, ймовірно, частково компенсує відсутність або нестачу білка дистрофіну, який спостерігається при різних видах м'язової дистрофії. Перед тим, як ці препарати використовуватимуться в лікуванні людей, ученим ще належить досліджувати їхню безпеку та ефективність. В людини є білок міостатин (myostatin), який обмежує м'язовий ріст. Дослідники відзначили покращення стану м'язів мишей, хворих на м'язову дистрофію Дюшена, після блокування цього білка. Біотехнологічна компанія працює над створенням препарату, який зможе блокувати міостатин у мишей, та планує подальші тести, які б дозволили використовувати цю технологію при лікуванні різних видів м'язової дистрофії у людей.
М'язова дистрофія - ряд м'язових захворювань, що характеризуються ослабленням та атрофією різних м'язів, зазвичай ці захворювання є спадковими. Загальні симптоми атрофії м'язів, видимі неозброєним оком - поганий чи сповільнений розвиток моторики, низьке зростання, сильне викривлення хребта вперед, прогинання коліна ззаду (лат. Genu recurvatum) тощо. псевдогіпертрофія (слабкі м'язи при здається хорошому розвитку, м'язи збільшені). Відбувається загибель м'язових волокон із заміщенням їх жирової та сполучної тканиною, тому їх форма залишається тривалий час незмінною або навіть може здаватися, що людина інтенсивно займається спортом.
Симптоми
- Зниження м'язового тонусу у уражених м'язах.
- Порушення розвитку моторики.
- Дитина починає ходити перевалку.
- Лордоз поперекового відділу.
- Низький зріст.
- Прогинання коліна ззаду.
- Збільшення жирової та сполучної тканини у м'язах.
Причини виникнення
Це спадкове захворювання. Деякі його форми схильні виключно до хлопчиків. Справжня причина м'язової дистрофії поки що невідома. Передбачається, що основним фактором захворювання є порушення обміну речовин у м'язах.
Лікування
Перед прогресуючою м'язовою дистрофією сучасна медицина безсила. Основна мета реабілітаційного лікування - підтримання фізичної активності пацієнта та покращення якості його життя, уповільнення процесу та відстрочення настання інвалідності. Хворим призначають вправи для тренування м'язів. Іноді застосовують клітинну терапію та призначають гормони. Також використовують різні шини, що перешкоджають утворенню контрактур.
У зв'язку з тим, що спадкове захворювання, профілактика допомогти не може. Підвищення кількості певних ферментів у крові вагітної означає велику ймовірність успадкування хвороби. Так як м'язова дистрофія - вкрай рідкісне захворювання, то майбутній мамі, в крові якої підвищена концентрація певних ферментів, навряд чи слід заздалегідь турбуватися. Дуже важливими є профілактичні огляди дітей.
До педіатра слід звернутися, якщо розвиток моторики у дитини повільніше, ніж у його однолітків, якщо йому важко вставати, підніматися сходами, якщо є скарги на слабкість і біль у ногах. За інших форм атрофії м'язів уражаються лицьові м'язи. Тому, якщо дитині важко заплющити очі, підняти руку або витягнути губи вперед, слід звернутися до лікаря.
Насамперед лікар ґрунтовно обстежує маленького пацієнта. При підозрі на дистрофію м'язів виконає електроміографію (ЕМГ), під час якої вимірювальний прилад зафіксує порушені реакції м'язів. Також він візьме шматочок тканини для мікроскопічного дослідження з метою виявлення певних змін. Крім того, візьме із вени кров на аналіз для визначення активності м'язових ферментів.
Перебіг хвороби
Перебіг різних м'язових дистрофій по-різному. Зазвичай захворювання починається непомітно, коли дитина починає ходити. Характеризується симетричним ураженням та слабкістю певних груп м'язів: спочатку уражаються м'язи обличчя, таза, плечового пояса або верхніх та нижніх кінцівок. Якщо дитина хвора на швидко прогресуючу форму дистрофії, то її здатність виконувати ті чи інші рухи швидко пропадають. До 8-10 років дитина так слабшає, що її доводиться возити в інвалідному візку. Інші форми атрофії м'язів прогресують повільніше, іноді з віком стан хворого може покращати. Однак навіть у цих випадках, будучи дорослими, хворі не можуть виконувати деякі рухи.
Існують такі форми дистрофії, у яких захворювання вперше виникає у зрілому віці. Згодом хворий, можливо, не зможе виконувати деякі фізичні дії, проте його життю небезпека не загрожує.
При бажанні мати дитину та наявності в сім'ї хворої на дистрофію серцевого м'яза Вам необхідно звернутися до центру генетики. Лікарі-генетики визначать можливість повторення цього захворювання.
М'язова дистрофія – це група спадкових захворювань, у яких м'язова маса та її функції поступово знижуються.
Різновиди м'язової дистрофії
Дев'ять типів м'язової дистрофії включають:
М'язова дистрофія Дюшенна (МДД) що впливає на хлопчиків, викликаючи прогресивну м'язову слабкість, і зазвичай починається з ніг. Це найважча форма м'язової дистрофії.
М'язова дистрофія Беккера (БМД) , яка вражає старших хлопчиків та молодих чоловіків, м'якша за МДД.
М'язова дистрофія Емері-Дрейфуса (ЕДМД) , що впливає на хлопчиків, викликаючи контрактури і слабкість в литок, слабкість у плечах та верхній частині рук, а також вади провідності серця. Жінки з ЕДМД схильні до ризику серцевого блоку.
Лімб-поясна м'язова дистрофія (ЛГМД) , яка починається в пізньому дитинстві, в ранньому дорослому віці, і вражає як чоловіків, так і жінок, викликаючи слабкість у м'язах навколо стегон та плечей. Це найпостійніша форма м'язової дистрофії, і поділяється на кілька різних видів. Багато людей з підозрою на ЛГМД, ймовірно, були неправильно діагностовані у минулому; і таким чином поширеність захворювання важко оцінити.
Плече-лопатково-лицьова міопатія (ФСГ) , відома також як хвороба Ландузі-Дежеріна, яка починається в пізньому дитинстві, в ранньому дорослому віці, і вражає як чоловіків, так і жінок, викликаючи слабкість у м'язах обличчя, плечей та передпліч. Стегна та ноги також можуть бути порушені.
Міотонічна дистрофія (МД) , відома також як хвороба Штейнерта, яка зачіпає як чоловіків, так і жінок, внаслідок чого загальна слабкість зачіпає обличчя, ноги та руки та супроводжується нездатністю розслабити уражені м'язи (міотонія). Симптоми можуть розпочатися будь-коли від народження до зрілого віку.
Окулофарингеальна м'язова дистрофія (ОФМД) , що впливає на дорослих обох статей, викликаючи слабкість у м'язах очей та горла.
Дистальна м'язова дистрофія (ДД) , яка починається в середньому віці або пізніше, викликаючи слабкість у м'язах рук та ніг.
Вроджена м'язова дистрофія (КМД) , яка присутня від народження, призводить до генералізованої слабкості, і зазвичай прогресує повільно. Її підтип, який називається Фукуяма КМД, також включає розумову відсталість. Обидва захворювання трапляються вкрай рідко.
Причини та симптоми дистрофії м'язів
Деякі форми м'язової дистрофії, включаючи МДД, МДБ, КМД та більшість форм ЛГМД, обумовлені дефектами генів комплексу м'язових білків. Дефекти в білках комплексу призводять до дистрофії м'язів, які поступово вичерпують свою здатність до самовідновлення. МДД та БМД викликані дефектами в гені білка під назвою дистрофін. Відмінності між іншими захворюваннями менш зрозумілі.
 М'язова дистрофія є генетичним захворюванням, тобто викликана дефектами в генах. Гени, які пов'язані один з одним у хромосомах, мають дві функції. Вони кодують виробництво білків і є матеріалом успадкування. Батьки передають своїм дітям через гени повний набір інструкцій для створення власних білків.
М'язова дистрофія є генетичним захворюванням, тобто викликана дефектами в генах. Гени, які пов'язані один з одним у хромосомах, мають дві функції. Вони кодують виробництво білків і є матеріалом успадкування. Батьки передають своїм дітям через гени повний набір інструкцій для створення власних білків.
Оскільки обидва батьки передають генетичний матеріал дитині, останній несе дві копії кожного гена, по одному від кожного батька. При деяких захворюваннях обидві копії повинні мати недоліки. Такі захворювання називаються аутосомно-рецесивними. Деякі форми ЛГМД та ДД демонструють цей зразок успадкування, як і КМД. Людина тільки з однією некоректною копією називається носієм і не матиме захворювання, але може передати некоректний ген своїм дітям.
Інші захворювання виникають, коли лише одна копія гена зіпсована. Такі захворювання називаються аутосомно-домінантними. Цей зразок успадкування демонструють ЛГМД, ДМ, ФСП, ОФМД та деякі форми ДД.
Всі типи дистрофії м'язів відзначені м'язовою слабкістю як основний симптом. Розподіл симптомів, вік, початок хвороби та її прогрес істотно різняться. Біль також є симптомом дистрофії м'язів, як правило, через наслідки ослаблення.
Діагностика м'язової дистрофії
Діагностика м'язової дистрофії включає ретельне вивчення медичної історії пацієнта і ретельне медичне обстеження. Сімейна історія може дати важливі підказки, оскільки всі види дистрофії м'язів є генетичними.
Лабораторні діагностичні тести дистрофії м'язів можуть включати наступне:
 Рівень м'язового ферменту креатинкінази (CK) у крові.
Рівні CK зростають через пошкодження м'язів і можуть розглядатися в деяких випадках навіть до появи симптомів.
Рівень м'язового ферменту креатинкінази (CK) у крові.
Рівні CK зростають через пошкодження м'язів і можуть розглядатися в деяких випадках навіть до появи симптомів.
М'язова біопсія , у якій невеликий шматочок м'язової тканини видаляють на дослідження. Зміни у структурі м'язових клітин та наявність фіброзної тканини чи інших аберантних структур характерні для різних форм м'язової дистрофії. М'язова тканина також може бути досліджена на присутність або відсутність конкретних білків, у тому числі дистрофіну.
Електроміограма (ЕМГ) . ЕМГ використовується вивчення реакції м'язів на стимуляцію. Знижена реакція проявляється у м'язовій дистрофії.
Генетичні тести . Деякі типи м'язової дистрофії можуть бути ідентифіковані шляхом тестування на присутність мутантного гена.
Точні генетичні тести для МДД, МДБ, ДМ, кількох форм ЛГМД та ЕДМД.
Інші специфічні тести по мірі необхідності. При підозрі на ЕДМД та МДБ, наприклад, для тестування функції серця може знадобитися електрокардіограма.
У випадку більшості форм м'язової дистрофії, точний діагноз не складно встановити. Проте існують і винятки. Навіть з біопсією може бути складно провести різницю між ФСГ та іншим захворюванням м'язів, поліміозитом. М'язова дистрофія у дітей початкової ЛГМД часто помилково сприймається набагато загальнішу МДД, особливо коли це відбувається у хлопчиків. БМД із раннім початком дуже схожа на МДД. М'язова дистрофія в дітей віком може бути помилково прийнята за одне із захворювань рухових нейронів, наприклад, спинально-м'язову атрофію; захворювання нервово-м'язової сполуки (міастенія); та інші захворювання м'язів.
Лікування дистрофії м'язів
 Власне, немає певних препаратів на лікування будь-якого з типів м'язової дистрофії. Преднізон та кортикостероїди показані для затримки прогресування МДД до певної міри. Преднізолон також призначають при БМД.
Власне, немає певних препаратів на лікування будь-якого з типів м'язової дистрофії. Преднізон та кортикостероїди показані для затримки прогресування МДД до певної міри. Преднізолон також призначають при БМД.
Лікування м'язової дистрофії в основному спрямоване на запобігання ускладненням, у тому числі зниження рухливості, спритності, контрактури, сколіозу, вад серця та дихальної недостатності.
Фізична терапія, зокрема, регулярне розтягування, використовується для підтримки діапазону рухів уражених м'язів і запобігання або затримки контрактури. Дужки використовуються частіше на кісточках та ногах. Зміцнення інших груп м'язів для компенсації слабкості може бути можливим, якщо уражені м'язи малі та ізольовані, а також на ранніх стадіях м'якших видів м'язової дистрофії. Регулярні вправи тим часом допомагають підтримувати загальний стан здоров'я. Серйозні фізичні навантаження, зазвичай, не рекомендуються.
Коли контрактури стають більш вираженими, то може бути виконана хірургічна операція.
Відмова від відповідальності:Інформація, подана в цій статті про дистрофію м'язів у дітей, призначена тільки для інформування читача. Вона може бути заміною для консультації професійним медичним працівником.
М'язова дистрофія- Захворювання м'язів, яке характеризується ослабленням скелетних м'язів, їх атрофією, часом до летального результату. Захворювання протікає без хворобливих відчуттів та симетрично по всьому тілу, чутливість не порушується. Дистрофія м'язів – генетичне захворювання.
Захворювання пояснюється тим, що організм не в змозі синтезувати та засвоювати необхідний білок для зростання м'язової тканини. Внаслідок цього м'язи стають слабкими і починають атрофуватися. Вони поступово перестають скорочуватися та розпадаються. На їхнє місце мали б утворюватися нові м'язові тканини, але з'являється лише жирова та сполучна тканина.
Види та класифікація
Вирізняють кілька видів м'язової дистрофії. По дії вони всі схожі, але відрізняються швидкістю розвитку захворювання та деякими аспектами його перебігу:
- Дистрофія Дюшенна- Виявляється дуже рано, в дитячому віці у дитини. Перші ознаки захворювання можуть виникнути, починаючи з 2 років життя малюка. Спочатку страждають м'язи нижніх кінцівок та попереку. Дитина починає погано ходити: перевалюється із боку на бік. Після цього взагалі втрачається здатність пересуватися, зазвичай до 10 років діти із захворюванням Дюшенна прикуті до інвалідного візка, оскільки м'язова тканина замінюється жировою. Після цього починається дистрофія інших м'язів. Часто до 15 років підлітки не здатні підняти руки вище за голову. Пацієнти з м'язовою дистрофією Дюшенна часто не живуть більше 20 років.
Профілактичних засобів немає, оскільки це захворювання викликане порушенням і мутацією генів.
- Захворювання Штейнерта.На цю недугу страждають люди у старшому віці (від 25 років), але зафіксовані випадки і в дитячому віці. М'язова дистрофія Штейнерта протікає досить повільно. Особливості цієї хвороби полягають у тому, що страждають як скелетні м'язи, а й лицьові, і навіть внутрішніх органів. Прожити з таким захворюванням можна досить довго (до 50-60 років), оскільки воно прогресує дуже повільно.
- Дистрофія Беккераза симптомами перебігу дуже схожа з дистрофією Дюшенна, але прогресує набагато повільніше. З таким захворюванням люди живуть довго. Страждають пацієнти в основному від супутніх травм та захворювань, адже сама хвороба Беккера протікає повільно. Зустрічається досить рідко.
- Захворювання Ерба-Ротазустрічається у підлітковому віці. М'язи починають атрофуватися з плечей, рук, а потім переходять на поперек та ноги. Хворому стає складно ходити. Хвороба прогресує повільно.
- Лікувально-лопатково-лицьова форма дистрофіїможе проявитися у будь-якому віці. Захворювання вражає м'язи обличчя та плечей: неможливо зімкнути губи та згорнути їх у трубочку, неможливо повністю зімкнути повіки. Захворювання протікає повільно, що не погіршує працездатність та нормальну життєдіяльність упродовж багатьох років. Але після 20-25 років розвитку захворювання починають атрофуватися м'язи кінцівок, що впливає на нормальне пересування, ускладнюючи його.
Причини та симптоми
Причини виникнення м'язової дистрофії:
- М'язова дистрофія – генетичне захворювання.
- Причинами цього захворювання є мутація тих чи інших генів, які відповідають за правильний синтез білка в організмі.
- Кожен вид дистрофії спричинений мутацією окремих генів.
Симптоми перебігу м'язової дистрофії
Залежно від виду м'язової дистрофії симптоми можуть відрізнятися, але все ж таки вони мають загальний характер:
- Слабкі руки та ноги.
- Зміна ходи чи втрата здатності ходити зовсім.
- Часті травми та падіння.
- Відсутність болючих відчуттів.
- Постійна втома.
- Втрата багатьох набутих навичок: ходьба, сидіння, підняття рук та ін.
Лікування м'язової дистрофії
Загальні відомості:
- Це захворювання є невиліковним, тому на сьогоднішній день немає препаратів, які можуть вилікувати дистрофію.
- Прогресуюча. Основною метою є уповільнення прогресуючої дистрофії, що досягається за допомогою спеціальних фізіотерапевтичних процедур.
- Хворим призначають лікувальні масажі та по можливості фізкультуру.
- Всі ці призначення індивідуальні, залежать від ступеня розвитку та тяжкості існуючої дистрофії.
- Хворим призначають внутрішньом'язові ін'єкції таких препаратів, як вітамін В1 та кортикостероїдів.
- Це речовини, які необхідні для правильної роботи та розвитку м'язів.
- Вони допомагають зберегти м'язову тканину.
- Захворювання не лікуватися, але правильний підхід дозволить полегшити його протікання та збільшити працездатність та нормальну життєдіяльність.
Можливі ускладнення
Нерідко у людей з м'язовою дистрофією виникають ускладнення, ось деякі з них:
- Проблеми з хребтом: деформація, біль.
- Інвалідність.
- Захворювання серця та дихальної системи.
- Порушення інтелектуальних здібностей та ін.
Пацієнтам при м'язовій дистрофії призначають лікарські засоби для полегшення симптомів. Ці ліки не здатні вилікувати захворювання, але вони стимулюють ріст м'язової тканини. Деякі з них:

У сучасній неврології існує безліч захворювань, характер виникнення яких не піддаються раціональному поясненню з боку фахівців. До таких хвороб можна віднести групу таких недуг, як дистрофія м'язів. Усього різновидів цієї хвороби дев'ять, але про все по порядку.
М'язова дистрофія – це хронічне спадкове захворювання внаслідок розвитку якого відбувається поразка м'язової системи людини. Уражені м'язи перестають нормально функціонувати, стоншуються в розмірах, а в місці їх розташування в організмі поступово наростає жировий прошарок.
Різновиди м'язової дистрофії
У сучасній неврології ця недуга класифікується на дев'ять різних хвороб. Класифікація захворювання пов'язана з:
- локалізацією м'язових порушень;
- характеристиками хвороби;
- агресивністю розвитку;
- віком.
Так, м'язова дистрофія буває:
- Дюшенна;
- міотонічна (хвороба Штейнерта);
- Бекера;
- Ерба Рота;
- юнацька форма дистрофії Ерба-Рота;
- плече – лопаткова лицьова форма (Ландузі-Дежеріна);
- алкогольна міопатія;
- дистальна форма;
- міодистрофія Емері-Дрейфуса.
Дистрофія Дюшенна
Найбільш відома форма прогресуюча дистрофія Дюшенна (псевдогіпертрофічна дистрофія, мерозин – негативна вроджена дистрофія). Цей різновид недуги проявляється у дитячому віці починаючи з двох до п'яти років. Насамперед від цієї недуги страждають м'язи нижніх кінцівок, які незважаючи на малорухливий спосіб життя у маленьких пацієнтів, поступово збільшуються у розмірах. Ця особливість пов'язана з наростанням жирової тканини замість м'язів.

Зліва здорова дитина, справа хвора
Поступово, у міру прогресування хвороби, вона переходить у верхню частину і вражає м'язи верхніх кінцівок. Як правило, вже до дванадцятирічного віку маленький пацієнт перестає повністю рухатися. Летальність у захворювання дуже висока приблизно 85-90% хворих не доживають до 20-річного віку.
У групі ризику знаходяться представники чоловічої статі, оскільки дівчаток ця недуга практично не торкається.
Хвороба Штейнерта
Природжена дистрофія Штейнера недарма має назву міотонічна, тому що в результаті прогресування хвороби відбувається надто повільне розслаблення м'язів після їхнього скорочення (таке явище називається міотонією).
Дане захворювання, на відміну від попереднього, поширене у дорослих, у віці від 20 до 40 років. Мають місце і випадки прогресування хвороби у дітей, як правило, у дитячому віці, однак, це швидше виняток, ніж правило.

На особу ознаки міотонії (відкритий рот і повіки)
Гендерної залежності хвороба не має і однаково від неї страждають як чоловіки, так і жінки. Зазначається, що з недузі проявляється слабкість мімічних м'язів обличчя, і навіть кінцівок. Прогресування, на відміну хвороби Дюшенна відбувається повільно.
Відмінна риса захворювання, можливість ураження як м'язів кінцівок, а й внутрішніх м'язів (серцевий м'яз), що, своєю чергою, становить велику небезпеку життя людини.
Хвороба Бекера
Ця форма хвороби також довго прогресує, і пацієнт тривалий час почувається добре. Загострення недуги може статися і натомість отриманих травм чи різних захворювань нервової системи, які своєю течією прискорять розвиток недуги.
У групі ризику є люди низького зростання.
Хвороба Ерба-Рота та юнацька її форма
Дане аутосомно – рецесивне захворювання розвивається у пацієнтів після 20 років, а юнацька форма у дітей та підлітків 11-13-річного віку. Прогресування хвороби відбувається за висхідним варіантом, тобто спочатку уражаються м'язи нижніх кінцівок, і поступово відбувається сходження недуги до верхніх кінцівок. 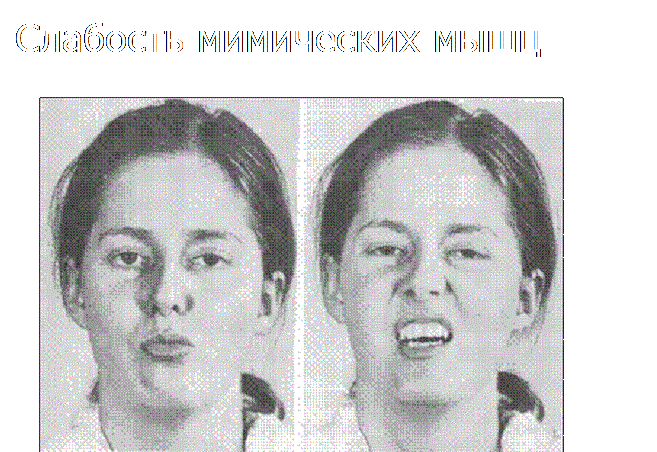
Відмінною рисою недуги є наявність лопаток, які в міру прогресування виступають більш явно і очевидно.
Під час ходьби відзначається перевалювання пацієнта, випинання живота та засув грудної клітки.
Хвороба Ландузі-Дежеріна
Плече-лопаточно-лицьова форма даного захворювання є найбільш безрозбірним видом, оскільки вражає людей віком від п'яти до 55 років. Для цієї недуги характерний дуже тривалий розвиток, пацієнт може зберігати працездатність до 25 років життя з цією хворобою.
Відмінними симптомами є ураження м'язів обличчя, внаслідок чого у хворого можуть виникнути проблеми з чіткістю вимови, у зв'язку з неповним змиканням губ. Крім того, відзначається неповне змикання повік.
У міру розвитку у хворого атрофуються лицьові м'язи, м'язи плечей, кінцівки та тулуба.
Ця форма залежить від генетичної мутації.
Алкогольна міопатія
Даний вид хвороби також не пов'язаний із генними мутаціями і причина його виникнення одна – зловживання алкоголем. У хворих можуть відзначати больові синдроми в кінцівках, пов'язані саме з ураженням м'язів.
Розрізняють гостру та хронічну алкогольну міопатію.
Дистальна форма
Дистальна форма м'язової дистрофії є доброякісним варіантом прогресуючої дистрофії. Як правило, це захворювання важко диференціювати від невральної аміатрофії Марі-Шарко. Для поділу цих двох захворювань потрібно проведення електроенцефалограми голови, яка і дає розуміння з якою хворобою має бути справа.
До основних симптомів недуги відносять атрофію м'язів кінцівок з їх подальшим схудненням. Можливі парези стоп, кистей рук тощо.
Міодистрофія Емері-Дрейфуса
Даний тип хвороби спочатку взагалі не був виділений в якості окремого захворювання, так як за своєю симптоматикою був схожий з дистрофією Дюшенна. Однак, надалі в результаті тривалого дослідження було встановлено, що хвороба Емері-Дрейфуса має індивідуальну симптоматику.
Хворобу відносять до розряду рідкісних. У групі ризику перебувають люди до 30 років, зазвичай, молодий вік. Є свідчення про прояв симптомів і після 30 років, але вони поодинокі.
Основна відмінність цього виду хвороби - це проблеми з м'язами серця, які можуть стати причиною смерті. Кардіоміопатія при цій недузі не єдина відмінність. Крім проблем із серцем, у пацієнтів спостерігають стандартні для дистрофії Дюшенна ознаки, але в більш щадному режимі розвитку.
Причини
Негативна складова хвороб нервової системи у тому, що вони важко піддаються вивченню. З цієї причини не до кінця відомо про фактори, які провокують розвиток тієї чи іншої форми м'язової дистрофії.
Основна причина для формування більшості підвидів даної хвороби є генна мутація, зокрема гена, який відповідає за синтез білка.
Наприклад, захворювання Дюшенна має пряме відношення до мутації статевої Х хромосоми. Основний переносник негативного гена виступають дівчата, які незважаючи на його наявність у своїй ДНК не страждають від цієї хвороби.
Що стосується міотонічної форми, то винуватець її виникнення ген, розташований у 19 хромосомі.
Основні симптоми
Наявність великої кількості різних підвидів у цієї недуги вказує на відмінності в симптоматиці, однак у хвороби є загальні симптоми, які включають:

Діагностика м'язової дистрофії
Діагностичні заходи при такій недузі великі, оскільки існує велика кількість хвороб, від яких необхідно диференціювати хворобу.
На початковому етапі лікар обов'язково вивчить анамнез хворого, уточнить про симптоматику, режим дня і т. п. Ці дані потрібні для складання плану проведення наступних діагностичних заходів, які можуть включати:

Чим пізніше проявилася хвороба, тим краще для хворого, оскільки ранні симптоми здебільшого закінчуються смертю.
Лікування
Лікування м'язової дистрофії - процес складний і затяжний, проте, в даний час не створено ліки, які повністю зцілюють хворого. Усі заходи спрямовані на полегшення життя хворого та відновлення деяких втрачених здібностей.
Для загальмовування розвитку хвороби призначають такі препарати:
- кортикостероїди;
- вітаміни В1;
- аденозинтрифосфат (АТФ).
Крім того, для уповільнення процесу розвитку використовують фетальні стволові клітини, які уповільнюють процес дистрофії.
Крім того, як профілактичні заходи призначають:
- масаж;
- фізіотерапію;
- дихальну гімнастику.
Крім стандартних варіантів лікування важливо постійно керуватися трьома головними складовими в процесі життєдіяльності:
- Адекватна фізична активність.
- Своєчасна психологічна підтримка.
- Дотримання дієти.
Фізична активність
Відсутність бажання в людини боротися з недугою негативно впливає на організм. Судіть самі, пасивність, небажання рухатися пригнічує і так уражену м'язову систему. М'язам необхідно давати навантаження, тому що без навантаження дистрофічні процеси починають відбуватися швидше, тим самим прогресуючи у швидшому темпі.
Помірна фізична активність, використання підтримувальних пристроїв будуть чудовою підмогою у боротьбі з недугою.
За наявності болю в м'язах чудово підійде плавання, йога, вправи на розтяжку.
Психологічна підтримка
Психологічна підтримка оточення є важливою для хворої людини. А якщо недуга така серйозна, як ця тим більше. Для когось буде достатньо звичайної підтримки з боку друзів та рідних, а комусь може знадобитися кваліфікована психологічна допомога.
Важливо дати зрозуміти такій людині, що вона не залишилася наодинці зі своєю проблемою. Він повинен розуміти, що йому є до кого звернутися, є люди, які співпереживають та підтримують його.
Дієта
Що стосується режиму харчування та дотримання дієти, існує думка, що дотримання протизапальної дієти може уповільнити прогресування недуги. Ця дієта знижує запалення, рівень глюкози, виводить токсини з організму та живить його корисними речовинами.
Суть такої дієти наступного:
- Відмова від продуктів, що містять «погані» жири та заміна їх на «хороші», введення в раціон ненасичених жирів, які містяться в оливковій, лляній, кунжутній олії, авокадо.
- Застосування в їжу м'яса та риби, при виробництві яких були використані антибіотики чи гормони.
- Повне видалення з раціону рафінованого цукру та глютену.
- Вживання в їжу наступних продуктів - китайська капуста, броколі, селера, ананас, лосось, буряк, кукумарія, імбир, куркума та інших продуктів, що мають протизапальні властивості.
- Молочні продукти допустимі лише на основі овечого та козячого молока.
- Допустимо вживання трав'яного чаю, лимонаду, квасу, морсів і натуральних соків.
Профілактика
Оскільки м'язову дистрофію досить важко передбачити та виявити на ранньому етапі, профілактичні заходи зводяться до двох простих рекомендацій:
Обов'язкове обстеження жінки на етапі планування вагітності щодо наявності мутацій у генах
Якщо до вагітності, з якихось причин не було зроблено генетичного обстеження, вже в період вагітності проводиться тест на визначення у плода мутацій у Х хромосомі.
Прогноз та ускладнення
Залежно від виду хвороби прогноз може бути різним, проте можна виділити кілька можливих ускладнень, що виникають при даній недузі.
- порушення серцевої діяльності
- викривлення хребта
- зниження інтелектуальних здібностей хворого
- зниження рухової активності
- порушення дихальної системи
- летальний результат
Отже, м'язова дистрофія є досить небезпечним і невиліковним захворюванням, тому майбутнім батькам необхідно з глибокою відповідальністю ставитися до планування вагітності. Бережіть себе та своїх майбутніх дітей!