Лекарства от дистрофии мышц. Прогрессирующие мышечные дистрофии. Редкие формы Х-сцепленных миодистрофий
![]()
Описание:
Мышечная дистрофия представляет собой группу хронических наследственных заболеваний скелетных мышц человека, проявляющихся слабостью и дегенерацией мышц. Существует девять различных форм мышечной дистрофии. Они различаются по таким характеристикам, как возраст, в котором начинается заболевание, локализация пораженных мышц, выраженность мышечной слабости, скорость прогрессирования дистрофии и тип ее наследования. Чаще всего встречаются две формы: мышечная дистрофия Дюшенна и миотоническая мышечная дистрофия.
Симптомы:
Дюшенна дистрофия. Х-хромосомная рецессивная мутация дистрофин-гена. Клинические признаки: начало в возрасте до 5 лет; прогрессирующая слабость мышц тазового и плечевого пояса; неспособность ходить после 12 лет; кифосколиоз; дыхательная недостаточность в возрасте 20-30 лет. Вовлечение других систем органов: ; снижение интеллекта.
Беккера дистрофия. Х-хромосомная рецессивная мутация дистрофин-гена. Клинические признаки: начало в раннем или позднем возрасте; медленно прогрессирующая слабость мышц тазового и плечевого пояса; сохранение способности ходить после 15 лет; дыхательная недостаточность после 40 лет. Вовлечение других систем органов: кардиомиопатия.
Миотоническая дистрофия. Аутосомно- доминантный; расширение нестабильного участка ДНК хромосомы 19ql3,3. Клинические признаки: начало в любом возрасте; медленно прогрессирующая слабость мышц век, лица, шеи, дистальных мышц конечностей; миотония. Вовлечение других систем органов: нарушение сердечной проводимости; психические нарушения; , лобная ; гонад.
Плече-лопаточно-лицевая дистрофия.
Аутосомно-доминантный; часто мутации хромосомы 4q35. Клинические признаки: начало в возрасте до 20 лет; медленно прогрессирующая мышечная слабость лицевой области, плечевого пояса, тыльного сгибания стопы. Вовлечение других систем органов: гипертензия; глухота.
Плечевого и тазового пояса (возможны несколько заболеваний). Аутосомно-рецессивный или доминантный. Клинические признаки: начало с раннего детства до среднего возраста; медленно прогрессирующая слабость мышц плечевого и тазового пояса. Вовлечение других систем органов: кардиомиопатия.
Глазо-глоточная дистрофия. Аутосомно-доминантный (Французская Канада или Испания). Клинические признаки: начало в 50-60 лет; медленно прогрессирующая слабость мышц: наружных глазных, век, лица и глотки; крикофарингеальная ахалазия. Вовлечение других систем органов: церебральные, глазные.
Врожденная дистрофия. Включает несколько заболеваний, в том числе типы Фукуяма и церебро-окулярная дисплазия). Аутосомно-рецессивный. Клинические признаки: начало при рождении; гипотония, задержка развития; в одних случаях - ранняя дыхательная недостаточность, в других - более благоприятное течение болезни.
Причины возникновения:
Заболевание вызывается аутосомно-доминантным геном с резко варьирующей экспрессивностью (возможность риска передачи на родственников 1-й степени составляет 50%). Заболевание вызывается амплификацией, т. е. повышением числа CTG - триплетов в определенном локусе 19 хромосомы (тип 1 миотонической дистрофии) или CCTG в 3 хромосоме (тип2 миотонической дистрофии). Тип 2 миотонической дистрофии изучен слабо. Считается, что он встречается только в 2% случаев (но может быть и значительно чаще); не взаимосвязан с типом 1; скорее всего не является причиной врожденных форм дистрофии, когда носителем является мать. Для 1 типа доказано, что число повторов нуклеотидов увеличивается при передаче мутации из поколения в поколение. Тяжесть заболевания четко коррелирует с численностью этих повторов. Наибольшее их число определяется при врожденной тяжелой форме заболевания . Выявленный механизм объясняет феномен антиципации - утяжеления и все более раннего начала болезни в нисходящих поколениях. К примеру, если генетический анализ показал, что родитель имеет определенное число повторов CTG, то у его ребенка обнаружится еще большее количество повторов этого триплета.
Лечение:
На сегодняшний день не существует способов предотвратить или замедлить прогрессирование данного заболевания. Терапия направлена главным образом на борьбу с осложнениями, такими, как деформация позвоночника, развивающаяся вследствие слабости мышц спины, или предрасположенность к пневмониям, обусловленная слабостью дыхательных мышц. Фенитоин, прокаинамид, хинин применяются в лечении миотонии, но требуется осторожность у больных с заболеваниями сердца (опасность ухудшения сердечной проводимости). Имплантация водителя сердечного ритма необходима больным с синкопе или сердечной блокадой. При лечении сердечных нарушений рекомендован препарат фенигидин. Применение ортопедических аппаратов может укрепить «висячие» стопы, стабилизировать голеностопные суставы, уменьшить частоту падений. Хорошо подобранная тренировка также может оказать положительное влияние на течение данного заболевания. При наличии атрофии применяют анаболические стероиды (ретаболил, неробол), общеукрепляющую терапию. В тех случаях, когда имеется значительно выраженная миотоническая симптоматика, назначают курсы дифенина по 0,03-0,05 г 3 раза в день, длительностью 2-3 нед. Предполагают, что дифенин оказывает угнетающее действие на синаптическую проводимость и снижает посттетаническую активность в мышцах. При повышенной сонливости, нередко сопровождающей миотоническую дистрофию, положительный эффект наблюдается при приеме селегилина. Рекомендован также прием некоторых биологически активных добавок: кофермента Q10 (100 мг/ день), витамина Е (200 МЕ/день) и селена (200 мкг/день), лецитина (20 г/день).
Эффективное излечение от данного заболевания возможно только при помощи генной терапии, которая сейчас интенсивно развивается. Многочисленные эксперименты показывают улучшение состояния мышечных волокон при лечении некоторых форм мышечной дистрофии. При дистрофиях Дюшена и Беккера наблюдается недостаточная выработка мышечного белка дистрофина. Ген, ответственный за производство этого белка, является самым большим из всех известных генов, поэтому для проведения генной терапии ученые создали миниатюрную версию этого гена. Лучшими проводниками гена к мышцам ученые признали аденовирусы. Поэтому внутрь аденовируса они поместили нужный ген и ввели его мышам, страдающим от недостатка дистрофина. Результаты опыта оказались обнадеживающими. В других сходных работах носителями этого гена являются липосомы, микросферы, лактоферрин. Оригинальный подход к генотерапевтическому лечению МДД разрабатывается в Оксфордском университете группой под руководством Кей Девис (Kay Davies). Суть метода заключается в попытке дерепрессии аутосомного гомолога дистрофина – гена утрофина, продукт экспрессии которого мог бы быть способен компенсировать недостаток дистрофина во всех группах мышц. В эмбриогенезе человека приблизительно до семи недель развития дистрофин не экспрессируется и его функцию в мышцах выполняет белок утрофин. В промежутке между седьмой и 19 неделями развития экспрессируются оба белка и после 19 недели происходит замещение мышечного утрофина на дистрофин. После 19 недели эмбрионального развития утрофин обнаруживается только в области нервно-мышечных контактов. Белок утрофин, имея аутосомную локализацию разительно напоминает дистрофин своими N- и С- концевыми доменами, играющими решающую роль в функции дистрофина. Результаты экспериментов указывают на принципиальную возможность коррекции дефектов в мышечных волокнах лишенных дистрофина с помощью утрофина Установлено, что два препарата (L-arginine и heregulin) увеличивают производство белка атрофин в мышечных клетках мышей. Повышенное количество атрофина, вероятно, частично скомпенсирует отсутствие или недостаток белка дистрофин, который наблюдается при различных видах мышечной дистрофии. Перед тем, как эти препараты будут использоваться в лечении людей, ученым еще предстоит исследовать их безопасность и эффективность. В организме человека есть белок миостатин (myostatin), который ограничивает мышечный рост. Исследователи отметили улучшение состояния мышц мышей, больных мышечной дистрофией Дюшена, после блокировки этого белка. Биотехнологическая компания работает над созданием препарата, который сможет блокировать миостатин у мышей, и планирует дальнейшие тесты, которые позволили бы использовать эту технологию при лечении различных видов мышечной дистрофии у людей.
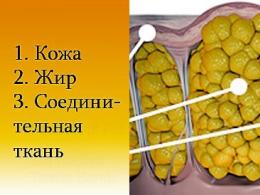
Мышечная дистрофия - ряд мышечных заболеваний, характеризующихся ослаблением и атрофией различных мышц, обычно эти заболевания являются наследственными. Общие симптомы атрофии мышц, видимые невооруженным глазом - плохое или замедленное развитие моторики, низкий рост, сильное искривление позвоночника вперед, прогибание колена сзади (лат. Genu recurvatum) и т.н. псевдогипертрофия (слабые мышцы при кажущемся хорошем развитии, мышцы увеличены). Происходит гибель мышечных волокон с замещением их жировой и соединительной тканью, поэтому их форма остается длительное время неизменной или даже может казаться, что человек интенсивно занимается спортом.
Симптомы
- Снижение мышечного тонуса в пораженных мышцах.
- Нарушение развития моторики.
- Ребенок начинает ходить вперевалку.
- Лордоз поясничного отдела.
- Низкий рост.
- Прогибание колена сзади.
- Увеличение жировой и соединительной ткани в мышцах.
Причины возникновения
Это наследственное заболевание. Некоторым его формам подвержены исключительно мальчики. Истинная причина мышечной дистрофии пока неизвестна. Предполагается, что основным фактором заболевания является нарушение обмена веществ в мышцах.
Лечение
Перед прогрессирующей мышечной дистрофией современная медицина бессильна. Основная цель реабилитационного лечения - поддержание физической активности пациента и улучшение качества его жизни, замедление процесса и отсрочка наступления инвалидности. Больным назначают упражнения для тренировки мышц. Иногда применяют клеточную терапию и назначают гормоны. Также используют различные шины, препятствующих образованию контрактур.
В связи с тем, что заболевание наследственное, профилактика помочь не может. Повышение количества определенных ферментов в крови беременной означает большую вероятность наследования болезни. Так как мышечная дистрофия - крайне редкое заболевание, то будущей маме, в крови у которой повышена концентрация определенных ферментов, вряд ли следует заранее беспокоиться. Очень важны профилактические осмотры детей.
К педиатру следует обратиться, если развитие моторики у ребенка более медленное, чем у его сверстников, если ему трудно вставать, подниматься по лестнице, если имеются жалобы на слабость и боли в ногах. При других формах атрофии мышц поражаются лицевые мышцы. Поэтому, если ребенку трудно закрыть глаза, поднять руку или вытянуть вперед губы, следует обратиться к врачу.
Прежде всего врач основательно обследует маленького пациента. При подозрении на мышечную дистрофию выполнит электромиографию (ЭМГ), во время которой измерительный прибор зафиксирует нарушенные реакции мышц. Также он возьмет кусочек ткани для микроскопического исследования с целью обнаружения определенных изменений. Кроме того, возьмет из вены кровь на анализ для определения активности мышечных ферментов.
Течение болезни
Течение различных мышечных дистрофий различно. Обычно заболевание начинается незаметно, когда ребенок начинает ходить. Характеризуется симметричным поражением и слабостью определенных групп мышц: сначала поражаются мышцы лица, таза, плечевого пояса или верхних и нижних конечностей. Если ребенок болен быстро прогрессирующей формой дистрофии, то его способности выполнять те или иные движения быстро пропадают. К 8-10 годам ребенок так ослабевает, что его приходится возить в инвалидной коляске. Другие формы атрофии мышц прогрессируют медленнее, иногда с возрастом состояние больного может улучшиться. Однако даже в этих случаях, уже будучи взрослыми, больные не могут выполнять некоторые движения.
Существуют такие формы дистрофии, при которых заболевание впервые возникает уже в зрелом возрасте. Со временем больной возможно не сможет выполнять некоторые физические действия, однако его жизни опасность не угрожает.
При желании иметь ребенка и наличии в семье больного дистрофией сердечной мышцы Вам необходимо обратиться в центр генетики. Врачи-генетики определят вероятность повторения этого заболевания.
Мышечная дистрофия – это группа наследственных заболеваний, при которых мышечная масса и ее функции постепенно снижаются.
Разновидности мышечной дистрофии
Девять типов мышечной дистрофии включают в себя:
Мышечная дистрофия Дюшенна (МДД) , которая влияет на мальчиков, вызывая прогрессивную мышечную слабость, и как правило, начинается с ног. Это самая тяжелая форма мышечной дистрофии.
Мышечная дистрофия Беккера (БМД) , которая поражает старших мальчиков и молодых мужчин, более мягкая, чем МДД.
Мышечная дистрофия Эмери-Дрейфуса (ЭДМД) , которая влияет на мальчиков, вызывая контрактуры и слабость в икрах, слабость в плечах и верхней части рук, а также пороки проводимости сердца. Женщины с ЭДМД подвержены риску сердечного блока.
Лимб-поясная мышечная дистрофия (ЛГМД) , которая начинается в позднем детстве, в раннем взрослом возрасте, и поражает как мужчин, так и женщин, вызывая слабость в мышцах вокруг бедер и плеч. Это самая непостоянная форма мышечной дистрофии, и подразделяется на несколько различных видов. Многие люди с подозрением на ЛГМД, вероятно, были неправильно диагностированы в прошлом; и таким образом, распространенность заболевания трудно оценить.
Плече-лопаточно-лицевая миопатия (ФСГ) , известная также как болезнь Ландузи-Дежерина, которая начинается в позднем детстве, в раннем взрослом возрасте, и поражает как мужчин, так и женщин, вызывая слабость в мышцах лица, плеч и предплечий. Бедра и ноги также могут быть затронуты.
Миотоническая дистрофия (МД) , известная также как болезнь Штейнерта, которая затрагивает как мужчин, так и женщин, в результате чего общая слабость затрагивает лицо, ноги и руки и сопровождается неспособностью расслабить пораженные мышцы (миотония). Симптомы могут начаться в любое время от рождения до зрелого возраста.
Окулофарингеальная мышечная дистрофия (ОФМД) , которая влияет на взрослых обоих полов, вызывая слабость в мышцах глаз и горла.
Дистальная мышечная дистрофия (ДД) , которая начинается в среднем возрасте или позже, вызывая слабость в мышцах рук и ног.
Врожденная мышечная дистрофия (КМД) , которая присутствует от рождения, приводит к генерализованной слабости, и обычно прогрессирует медленно. Ее подтип, который называется Фукуяма КМД, также включает в себя умственную отсталость. Оба заболевания встречаются крайне редко.
Причины и симптомы дистрофии мышц
Некоторые формы мышечной дистрофии, включая МДД, МДБ, КМД, и большинство форм ЛГМД, обусловлены дефектами генов комплекса мышечных белков. Дефекты в белках комплекса приводят к дистрофии мышц, которые постепенно исчерпывает свою способность к самовосстановлению. МДД и БМД вызваны дефектами в гене белка под названием дистрофин. Различия между другими заболеваниями менее понятны.
 Мышечная дистрофия является генетическим заболеванием, то есть она вызвана дефектами в генах. Гены, которые связаны друг с другом в хромосомах, имеют две функции. Они кодируют производство белков, и являются материалом наследования. Родители передают своим детям через гены полный набор инструкций для создания своих собственных белков.
Мышечная дистрофия является генетическим заболеванием, то есть она вызвана дефектами в генах. Гены, которые связаны друг с другом в хромосомах, имеют две функции. Они кодируют производство белков, и являются материалом наследования. Родители передают своим детям через гены полный набор инструкций для создания своих собственных белков.
Поскольку оба родителя передают генетический материал своему ребенку, последний несет две копии каждого гена, по одному от каждого родителя. При некоторых заболеваниях обе копии должны иметь недостатки. Такие заболевания называются аутосомно-рецессивными. Некоторые формы ЛГМД и ДД демонстрируют этот образец наследования, как и КМД. Человек только с одной некорректной копией, называется носителем, и не будет иметь заболевание, но может передать некорректный ген своим детям.
Другие заболевания возникают, когда только одна копия гена испорчена. Такие заболевания называются аутосомно-доминантными. Этот образец наследования демонстрируют ЛГМД, ДМ, ФСН, ОФМД и некоторые формы ДД.
Все типы дистрофии мышц отмечены мышечной слабостью в качестве основного симптома. Распределение симптомов, возраст, начало болезни и ее прогрессирование существенно различаются. Боль также является симптомом дистрофии мышц, как правило, из-за последствий ослабленности.
Диагностика мышечной дистрофии
Диагностика мышечной дистрофии включает в себя тщательное изучение медицинской истории пациента и тщательное медицинское обследование. Семейная история может дать важные подсказки, так как все виды мышечной дистрофии являются генетическими.
Лабораторные диагностические тесты дистрофии мышц могут включать в себя следующее:
 Уровень мышечного фермента креатинкиназы (CK) в крови.
Уровни CK растут из-за повреждения мышц и могут рассматриваться в некоторых случаях даже до появления симптомов.
Уровень мышечного фермента креатинкиназы (CK) в крови.
Уровни CK растут из-за повреждения мышц и могут рассматриваться в некоторых случаях даже до появления симптомов.
Мышечная биопсия , в которой небольшой кусочек мышечной ткани удаляют для исследования. Изменения в структуре мышечных клеток и наличие фиброзной ткани или других аберрантных структур характерны для различных форм мышечной дистрофии. Мышечной ткани также могут быть исследованы на присутствие или отсутствие конкретных белков, в том числе дистрофина.
Электромиограмма (ЭМГ) . ЭМГ используется для изучения реакции мышц на стимуляцию. Сниженная реакция проявляется в мышечной дистрофии.
Генетические тесты . Некоторые из типов мышечной дистрофии могут быть идентифицированы путем тестирования на присутствие мутантного гена.
Точные генетические тесты для МДД, МДБ, ДМ, нескольких форм ЛГМД и ЭДМД.
Другие специфические тесты по мере необходимости. При подозрении на ЭДМД и МДБ, например, для тестирования функции сердца может понадобиться электрокардиограмма.
В случае с большинством форм мышечной дистрофии, точный диагноз не сложно установить. Однако, существуют и исключения. Даже с биопсией может быть сложно провести различие между ФСГ и другим заболеванием мышц, полимиозитом. Мышечная дистрофия у детей начальной ЛГМД часто ошибочно принимается за гораздо более общую МДД, особенно когда это происходит у мальчиков. БМД с ранним началом очень похожа на МДД. Мышечная дистрофия у детей может быть ошибочно принята за одно из заболеваний двигательных нейронов, к примеру, спинально-мышечную атрофию; заболевания нервно-мышечного соединения (миастения); и другие заболевания мышц.
Лечение дистрофии мышц
 Собственно говоря, не существует определенных препаратов для лечения любого из типов мышечной дистрофии. Преднизон и кортикостероиды показаны для задержки прогрессирования МДД в некоторой степени. Преднизолон также назначают при БМД.
Собственно говоря, не существует определенных препаратов для лечения любого из типов мышечной дистрофии. Преднизон и кортикостероиды показаны для задержки прогрессирования МДД в некоторой степени. Преднизолон также назначают при БМД.
Лечение мышечной дистрофии в основном направленона предотвращение осложнений, в том числе снижение подвижности, ловкости, контрактуры, сколиоза, пороков сердца и дыхательной недостаточности.
Физическая терапия, в частности, регулярное растяжение, используется для поддержания диапазона движений пораженных мышц и для предотвращения или задержки контрактуры. Скобки используются чаще на лодыжках и ногах. Укрепление других групп мышц для компенсации слабости может быть возможным, если пораженные мышцы малы и изолированы, а также на ранних стадиях более мягких видов мышечной дистрофии. Регулярные упражнения, между тем, помогают поддерживать общее состояние здоровья. Серьезные физические нагрузки, как правило, не рекомендуются.
Когда контрактуры становятся более выраженными, может быть выполнена хирургическая операция.
Отказ от ответственности: Информация, представленная в этой статье про мышечную дистрофию у детей, предназначена только для информирования читателя. Она не может быть заменой для консультации профессиональным медицинским работником.
Мышечная дистрофия – заболевание мышц, которое характеризуется ослаблением скелетных мышц, их атрофией, порой до летального исхода. Заболевание протекает без болезненных ощущений и симметрично по всему телу, чувствительность не нарушается. Дистрофия мышц – генетическое заболевание.
Заболевание объясняется тем, что организм не в состоянии синтезировать и усваивать необходимый белок для роста мышечной ткани. В результате этого мышцы становятся слабыми и начинают атрофироваться. Они постепенно перестают сокращаться и распадаются. На их место должны были бы образовываться новые мышечные ткани, но появляется только жировая и соединительная ткань.
Виды и классификация
Выделяют несколько видов мышечной дистрофии. По действию они все схожи, но отличаются скоростью развития заболевания и некоторыми аспектами его протекания:
- Дистрофия Дюшенна – проявляется очень рано, в детском возрасте у ребенка. Первые признаки заболевания могут появиться, начиная с 2 лет жизни малыша. Сначала страдают мышцы нижних конечностей и поясницы. Ребенок начинает плохо ходить: переваливается с боку на бок. После и вовсе теряется способность передвигаться, обычно к 10 годам дети с заболеванием Дюшенна прикованы к инвалидной коляске, так как мышечная ткань заменяется жировой. После начинается дистрофия остальных мышц. Часто к 15 годам подростки не способны поднять руки выше головы. Пациенты с мышечной дистрофией Дюшенна зачастую не живут больше 20 лет.
Профилактических средств не существует, так как это заболевание вызванное нарушением и мутацией генов.
- Заболевание Штейнерта. Этим недугом страдают люди в старшем возрасте (от 25 лет), но зафиксированы случаи и в детском возрасте. Мышечная дистрофия Штейнерта протекает довольно медленно. Особенности этой болезни заключаются в том, что страдают не только скелетные мышцы, но и лицевые, а также внутренних органов. Прожить с таким заболеванием можно довольно долго (до 50-60 лет), так как оно прогрессирует очень медленно.
- Дистрофия Беккера по симптомам протекания очень схожа с дистрофией Дюшенна, но прогрессирует намного медленнее. С таким заболевание люди живут долго. Страдают пациенты в основном от сопутствующих травм и заболеваний, ведь сама болезнь Беккера протекает медленно. Встречается довольно редко.
- Заболевание Эрба-Рота встречается в подростковом возрасте. Мышцы начинают атрофироваться с плеч, рук, а затем переходят на поясницу и ноги. Больному становится сложно ходить. Болезнь прогрессирует медленно.
- Лече-лопаточно-лицевая форма дистрофии может проявиться в любом возрасте. Заболевание поражает мышцы лица и плеч: невозможно сомкнуть губы и свернуть их в трубочку, невозможно полностью сомкнуть веки. Заболевание протекает медленно, что не ухудшает трудоспособность и нормальную жизнедеятельность на протяжении многих лет. Но после 20-25 лет развития заболевания начинают атрофироваться мышцы конечностей, что влияет на нормальное передвижение, затрудняя его.
Причины и симптомы
Причины возникновения мышечной дистрофии:
- Мышечная дистрофия – генетическое заболевание.
- Причинами этого заболевания является мутация тех или иных генов, которые отвечаю за правильный синтез белка в организме.
- Каждый вид дистрофии вызван мутацией отдельных генов.
Симптомы протекания мышечной дистрофии
В зависимости от вида мышечной дистрофии симптомы могут отличаться, но все же они имеют общий характер:
- Слабые руки и ноги.
- Изменение походки или потеря способности ходить вовсе.
- Частые травмы и падения.
- Отсутствие болевых ощущений.
- Постоянная усталость.
- Потеря многих приобретенных навыков: ходьба, сидение, поднятие рук и др.
Лечение мышечной дистрофии
Общие сведения:
- Это заболевание является неизлечимым, поэтому на сегодняшний день нет препаратов, которые могут вылечить дистрофию.
- Прогрессирующая. Основной целью является замедления прогрессирующей дистрофии, что достигается с помощью специальных физиотерапевтических процедур.
- Больным назначают лечебные массажи и по возможности физкультуру.
- Все эти назначения индивидуальны, зависят они от степени развития и тяжести существующей дистрофии.
- Больным назначают внутримышечно инъекции таких препаратов, как витамин В 1 и кортикостероидов.
- Это вещества, которые необходимы для правильной работы и развития мышц.
- Они помогают сохранить мышечную ткань.
- Заболевания не лечиться, но правильный подход позволить облегчить его протекание и увеличить работоспособность и нормальную жизнедеятельность.
Возможные осложнения
Нередко у людей с мышечной дистрофии возникают осложнения, вот некоторые из них:
- Проблемы с позвоночником: деформация, боли.
- Инвалидность.
- Заболевания сердца и дыхательной системы.
- Нарушение интеллектуальных способностей и др.
Пациентам при мышечной дистрофии назначают лекарственные препараты для облегчения симптомов. Эти лекарства не способны излечить заболевания, но они стимулируют рост мышечной ткани. Некоторые из них:

В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.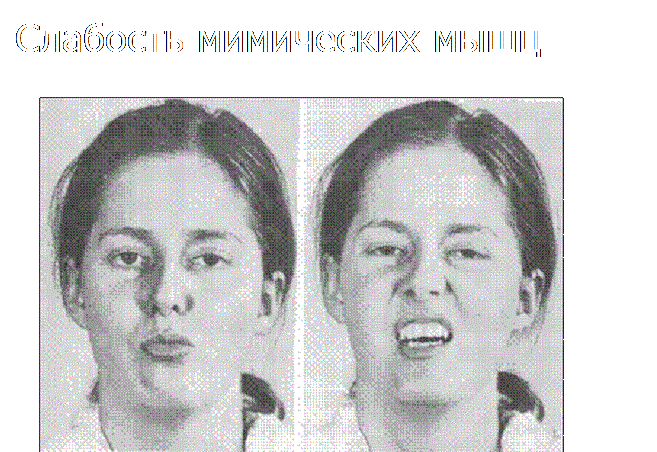
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!