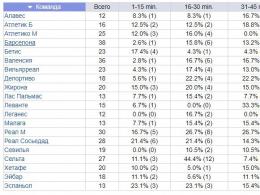
Neuromuskulaarsete haiguste sümptomid. Neuromuskulaarsed häired. Lihashaiguste tüübid
Iv. Vaptsarov
Lihashaigused on lapsepõlves suhteliselt tavalised. Mõned neist on tingitud lihaskiu esmasest kahjustusest. Need on kaasasündinud, geneetiliselt sõltuvad (pärilikud ja pärilikud-perekondlikud) haigused. Teised kujutavad endast metaboolsete häirete, nakkuslike, põletikuliste ja toksiliste protsesside tagajärjel tekkinud lihaskahjustusi. Kolmanda rühma haigused on põhjustatud närvisüsteemi ja neuromuskulaarse aparatuuri haigustest. Samuti on rühm, mis ühendab seni selgitamata etioloogiaga lihashaigusi.
ESMASED JA PÄRILIKUD LIHASHAIGUSED
PROGRESSIIVNE LIHASDÜSTROOFIA
Progresseeruvad lihasdüstroofiad on geneetiliselt määratud pärilikud ja pärilikud perekondlikud haigused, mida iseloomustab krooniline progresseeruv areng, mille tagajärjeks on raske puue. Nende haiguste suhteline esinemissagedus, mis viimasel ajal on suurenenud, kliiniliste sümptomite raskus, samuti spetsiifilise ja tõhusa ravi puudumine muudavad need sotsiaalseteks haigusteks.
Patogenees ja patoloogiline anatoomia. Primaarsete lihasdüstroofiate histoloogilist pilti iseloomustab ebaühtlane segmentaalne degeneratsioon, mis areneb piirkondades piki lihaseid. lihaskiud, mis nendes piirkondades kaotavad oma põikiriba. Sarcolemma tuumade suurus suureneb, need muutuvad ümaramaks ja asuvad keskpunktile lähemal. On pilt hüaliinsest, granulaarsest või vakuolaarsest düstroofiast, millel on iseloomulik kalduvus teatud määrdumisele. Nähtav fagotsütoos, kasv sidekoe ja rasvatilkade märkimisväärne kogunemine kiudude vahele, mis annab düstroofselt väljendunud lihasele kollaka värvuse. Eriti iseloomulik tunnus on aga kahjustuste juhuslik jaotus üksikutes kimpudes ja seetõttu on nende suurused erinevad. See omadus eristab progresseeruvaid lihasdüstroofiat närvilistest, kus eesmiste sarvede, juurte või tüve kahjustus põhjustab lihaskiudude süstemaatilist ja ühtlast, mitte segmentaalset atroofiat.
Nende düstroofiate patogeneetilisi mehhanisme ei ole veel täielikult välja selgitatud. Praegu on enim aktsepteeritud ensüümiteooria, mille kohaselt düstroofsed muutused lihaskiud tekivad lihaste aldolaasi, fosfokreatiinkinaasi ja vähemal määral laktaatdehüdrogenaasi aktiivsuse rikkumise tagajärjel. Haiguse esimestel faasidel nende ensüümide tase veres tõuseb, kuid paralleelselt atroofia progresseeruva arenguga väheneb neid tootva aktiivse lihaskoe vähenemise tõttu järk-järgult. Transaminaaside tase on tavaliselt normaalne. Hüperkreatinuuria ja hüpokreatinuuria tuvastatakse ka madala kreatineemiaga.
Elektromüogrammi iseloomustavad: a) elektrilise aktiivsuse puudumine puhkeolekus; b) mootoriüksuste madalad, kõverad ja mõnikord mitmefaasilised potentsiaalid; c) suureneva pingutusega häirekõverate kiire tekkimine; d) maksimaalse kontraktsiooni korral häirete salvestamise taustal kontrasteerub väljendunud lihasnõrkus.
Sõltuvalt haiguse geneetilise ülekande tüübist ja protsessi esialgsest lokaliseerimisest teatud lihasrühmades on üldiselt aktsepteeritud, et progresseeruvad lihasdüstroofiad esindavad mitmeid kliinilisi ja geneetilisi vorme.
Duchenne'i haigus(paralysis pseudohypertrophicans, paralysis myosclerotica) on X-seotud retsessiivne haigus, mis avaldub ligikaudu aasta pärast sündi ja peamiselt poistel. Seda iseloomustab kehatüve lihaste ja jäsemete proksimaalsete osade tugevuse üldine ja progresseeruv vähenemine ning jäsemete distaalsete osade lihaste suhteliselt säilinud motoorne jõud. Kõigepealt on kahjustatud alajäsemete lihased. Kõnnak omandab "pardi" iseloomu. Kõndimisel jääb keha maha kiiresti areneva lordoosi tõttu. Lapsed kukuvad sageli ja trepist üles ronimine on raske. Kui pärast kükitamist üritavad lapsed püsti tõusta, toetuvad nad peale. nende alajäsemed kätega vaheldumisi ümber paigutades.Kui laps lamab, siis püsti püüdes rullub ta end kõhuli, toetub kätele, painutab aeglaselt põlvi ja alles pärast seda sirgub, aidates. ise kätega, nagu eespool kirjeldatud.Siis katab kahjustus proksimaalseid lihaseid ülemised jäsemed ja õlavöötmega. Kaugelearenenud juhtudel põhjustab abaluulihaste atroofia mõnikord abaluu alatae ilmumist. Pärast suured lihased mõjutatud on väiksemad lihased. Kõikide jäsemete lihasrühmade sümmeetrilise, kuid ebaühtlase kaasatuse tõttu tekivad tugevad deformatsioonid ja selgroo kõverused. Nägu tavaliselt ei muutu. Mõnedes suuremates lihastes toimub koos kiudude atroofiaga sidekoe vohamine ja rasvade kogunemine, mille tulemuseks on pseudohüpertroofia, mis on Duchenne'i tõve iseloomulik tunnus. Seda protsessi väljendatakse kõige selgemalt nelipealihased, harvemini - deltalihases, mille mass on kontrastiks naaberlihaste atroofia taustal.
Reeglina jäävad kõõluste refleksid normaalseks, kuid tegelik lihaskontraktsioon nõrgeneb järsult.
Düstroofiline protsess võib mõjutada ka müokardit. Valkude ja rasvade degeneratsiooni ning fibroosi tagajärjel tekib kardiomegaalia. EKG näitab PQ laienemist, sageli ühe jala blokaadi ja T segmendi vähenemist. Pulss kiireneb ja lõppstaadiumis ilmnevad kardiovaskulaarse nõrkuse sümptomid. Atroofia ja liikumispiirangu tõttu täheldatakse osteoporoosi, "diafüüsi hõrenemist ja harvadel juhtudel luumurdu. Progresseeruv puue võib põhjustada muutusi iseloomus, kuid vaimse arengu mahajäämust täheldatakse harva.
Leideni haigus - Möbius on Duchenne'i tõve tüüp, mida iseloomustab pseudohüpertroofia puudumine ja protsessi lokaliseerimine ainult vaagna ja alajäsemete lihastes. See on pärilik - autosoomne retsessiivne tüüp.
Landouzy haigus - Dejerine nimetatakse Myopathia facio-scapulo-humeralis, kuna protsess algab näolihastest ja mõjutab valdavalt õlavöötme lihaseid. See pärineb autosomaalselt domineerival viisil ja mõjutab mõlemat sugupoolt võrdselt. Tavaliselt avaldub see teisel elukümnendil, kuid on kirjeldatud ka varasema ja hilisema alguse juhtumeid. Näolihaste düstroofia tulemuseks on iseloomulik müopaatiline nägu, millel on fikseeritud ilme, horisontaalne naeratus ja silmade mittetäielik sulgemine une ajal. Atroofia katab järk-järgult õlavöötme lihased (mm. dentatus, rhomboideus, trapezius, infra- et supraspinosus, m. m. pectorales, deltoideus, biceps et triceps brachiaiis jne), põhjustades liigutuste ja õla pterigoidse kuju olulise piirangu. labad (scapulae ala-tae) . Iseloomulik on pseudohüpertroofia puudumine enamikul patsientidel. Mõjutatud on ka müokard, kuid tavaliselt puuduvad kliinilised sümptomid ja diagnoos tehakse EKG abil. See haigusvorm areneb palju aeglasemalt, eksisteerib aastaid. Tema prognoos on hoolimata progresseeruvast puudest suhteliselt parem.
Erbi tõbi (Myopathia scapulo-humeralis) edastatakse autosoomselt domineerival viisil. Kliiniliste omaduste ja arengu poolest on see väga sarnane Landouzy-Dejerine'i haigusele, kuid erineb sellest näolihaste puudumise või hilise kahjustuse ning pseudohüpertroofia esinemise poolest.
Haruldased histoloogilised sordid
Duchenne'i tõve vastsündinu vorm sarnaneb kliiniliselt täielikult Oppenheimi tõvega (sündroom, mis varem ühendas nii primaarset kui ka neuraalset lihasdüstroofiat, vt Werdnig-Hoffmanni tõbi).
Primaarne kaasasündinud generaliseerunud Krabbe lihaste hüpoplaasia ja sellega seotud Batten-Turneni tõbi.
Keskne südamiku haigusmida iseloomustab müofibrillide rühmitus kimbu keskel ja ketta puudumine. Kliiniline pilt koosneb mittearenevast müotooniast, mis hiljem areneb selgelt väljendunud lihasnõrkuseni. See edastatakse autosomaalsel domineerival viisil.
Nemaliini müopaatiaon sarnane kliiniline pilt, kuid histoloogilisel uuringul leitakse Z-ketas, mille tropomüosiin moodustab sarkolemma all spetsiaalsed "pulgad".
Müotubulaarsed müopaatiad histoloogiliselt koosnevad nad loote tüüpi lihastest, mille kiu keskel on toruõõnsused, mis sisaldavad suurt hulka mitokondreid.
Mitokondriaalsed müopaatiad eristuvad mitmesugused mitokondriaalsed anomaaliad: kandmised, hiiglaslik suurus või ebatavaline suur kogus. Pärandi tüüp on autosoomne dominantne.
Progresseeruva haiguse diagnoosimine lihasdüstroofiaüksikasjaliku kliinilise pildi olemasolul on seda lihtne panna isegi esimesel läbivaatusel. Klassikaliste vormide eristamine on võimalik defineerimisega esialgne rühm kahjustused, pseudohüpertroofia olemasolu või puudumine ja anomaalia ülekandumise geneetiline tüüp.
Sellega seoses tuleb ette suuri raskusi varajased staadiumid haiguse areng, samuti ebatüüpiliste, kustutatud vormidega. Nendel juhtudel aitavad täpset diagnoosi panna perekonna geneetilised ja biokeemilised (ensüümi) uuringud.
Kogu rühma diferentsiaaldiagnostikas on vaja arvestada neuraalsete (Werdnig-Hoffmann, Kugelberg-Wellander) ja teiste sümptomaatiliste lihasdüstroofiate, müatoonia ja müotooniaga varajases eas jne.
Kliinik ja prognoos. Lihaste tagasitõmbumine ja kõõluste atroofia (nende vormide sümptomid) viivad järk-järgult kontraktuuride ja liigeste deformatsioonide tekkeni, mis kahjustavad lapse motoorseid funktsioone ja liigutusi. Teisest küljest kiirendab tegevusetus atroofiat, tekitades nõiaringi, mis lõpeb täieliku invaliidistusega. Duchenne'i, Leideni - Möbiuse ja Landouzy - Dejerine'i vormide prognoos halveneb progresseeruva müokardi düstroofia ja nende laste kalduvuse tõttu hingamisteede infektsioonidele. Progresseeruv puue mõjutab negatiivselt laste psüühikat, samas kui raskemate vormidega võib kaasneda teatav neuropsüühilise arengu viivitus. Sagedamini täheldatakse patoloogilisi muutusi iseloomus.
Narkootikumide ravi (adrenaliin, pilokarpiin, ezeriin, galaktamiin, nivaliin, proteolüsaadid, androgeensed anaboolsed hormoonid, E-vitamiin, glükokool, isegi adenosiintrifosforhape) ei anna märkimisväärseid tulemusi. Suuremal määral võite loota füsioteraapiale, mis parandab lihaste vereringet: soojad protseduurid, kerge massaaž jne. Samuti on ette nähtud vasodilataatorid, näiteks vasculat.
Täielik puhkus peegeldub ebasoodsalt. Laps peab tegema mõõdukaid, doseeritud aeglases rütmis liigutusi, mis ei põhjusta lihaste energiavarude ammendumist ega põhjusta seisundi halvenemist. Õige psühhopedagoogiline lähenemine on eriti vajalik oma haigust sügavalt põdevate laste meeleolu parandamiseks.
NÄRVISÜSTEEMI KAHJUSTUSEST PÕHJUSTATUD PÄRILIKU LIHASATROOFIA
Neuraalne lihasatroofia (Charcot-Marie-Toothi tõbi) - pärilik degeneratiivne haigus perifeerne närvisüsteem.
Spinaalne lihaste atroofia (Werdnig-Godfmanni tõbi). Selle haiguse peamine patoloogiline protsess on seljaaju eesmiste sarvede motoorsete rakkude progresseeruv degeneratsioon. Lihaste atroofia on sekundaarne nähtus.
Haiguse etioloogiat ei ole täielikult välja selgitatud.
Patoloogiline anatoomiline uuring tuvastab seljaaju eesmiste sarvede rakkude kahjustuse.
Kliinik. Haigus avaldub esimestel elupäevadel või esimestel kuudel. Tekib ebatavaliselt raske lihaste hüpotensioon, mis algab proksimaalsetest alajäsemetest ja levib kiiresti kogu skeletilihastesse. Laps lamab täiesti loidult, ilma toonuseta ja teeb väikseimate liigestega (näiteks sõrmedega) vaid kergeid liigutusi. Kuid näoilmete elavus vastandub teravalt jäsemete üldise loiduse ja lapse vaikse nõrga häälega. Passiivsed liigutused on võimalikud igas suunas ja liigesed jätavad mulje erakordsest lõtvusest. Hüpotensioon mõjutab järsult abihingamislihaseid, mistõttu hingamine ja kopsuventilatsioon on väga rasked. Sellest tuleneb atelektaatilise kopsupõletiku eriline sagedus ja hingamisteede infektsioonide tõsine kulg. Lihaste atroofia on väga väljendunud, kuid pilti varjab märkimisväärne nahaalune rasvkude. Röntgenpiltidel on aga lihaste hõrenemine selgelt näha. Pareesi ja halvatuse esinemine väljendub nõrgenemises või täielikus puudumises kõõluste refleksid olemasoleva taustal naha refleksid samuti tegelik lihaste kontraktsioon. Elektrilise erutuvuse uurimine paljastab kronaksia pikenemise ja lihaste degeneratsiooni reaktsiooni ning elektromüogramm neurogeense lihasatroofia.
Sellel haigusel on autosoomne retsessiivne ülekandemuster. See on tavaks jagada kolmeks peamiseks kliiniliseks vormiks: varajane (kaasasündinud), lapseea ja hiline (Kiegelberg-Welanderi tõbi). Viimasel ajal on kirjeldatud ka vahevorme.
Lülisamba lihaste atroofia varajane vorm avaldub juba emakasse loote puudumise või täiesti loid liigutustena, mis teeb muret eelkõige nendel rasedatel, kes on juba terve lapse sünnitanud.
Diagnoos tehakse kohe pärast sündi, kuna see jätab mulje teravast hüpotensioonist ja lapse liikuvuse vähenemisest. Tulevikus süvenevad hüpotensioon ja parees jätkuvalt. Lapse nägu kaotab täielikult oma näoilmed.
See haigusvorm langeb täielikult kokku Oppenheimi kirjeldatud kaasasündinud müotooniaga, mida ei ole hiljuti peetud iseseisvaks haiguseks, kuna nende haiguste üldised sümptomid võimaldasid ühendada need üheks nosoloogiliseks üksuseks.
Varaste vormide prognoos on raske. Lapsed surevad endiselt imikueas hingamisteede infektsioonidest. Hiliste ja kergete vormide korral, kui lapsed ei sure esimese kolme eluaasta jooksul, võib toimuda märkimisväärne kohanemine.
Ravi. Soovitatav on kasutada kõiki poliomüeliidi raviaineid. Kõige olulisem on aga hingamisteede infektsioonide ja nakkushaiguste ennetamine, millesse need lapsed tavaliselt surevad.
Clinical Pediatrics Toimetanud prof. Br. Bratinova
Neuromuskulaarsed haigused on pärilike ja mittepärilike haiguste rühm, mida iseloomustab düsfunktsioon:
- lihaste süsteem - müopaatia ja müotoonia;
- neuromuskulaarne sünaptiline aparaat - myasthenia gravis ja müasteenilised sündroomid;
- perifeersed närvid, seljaaju eesmiste sarvede motoorsed neuronid - sekundaarne (neurogeenne) amüotroofia (neuraalne ja seljaaju).
Neuromuskulaarsete haiguste rühma kuuluvad liikumisfunktsiooni häired ja lihasnõrkus. On järgmised neuromuskulaarsed haigused:
- müopaatia;
- müotoonia;
- sekundaarne (neurogeenne) amüotroofia;
- müasteenia.
NMZ diagnoosimine ja diferentsiaaldiagnostika, eriti kursuse algfaasis, on väga raske. Sellistel juhtudel suur tähtsus omandada geneetilisi, neurofüsioloogilisi, biokeemilisi ja morfoloogilisi uurimismeetodeid.
Neurofüsioloogilised uurimismeetodid:
- elektromüograafia (EMG);
- elektroneuromüograafia (ENMG).
Kohalik (nõel) EMG eriti informatiivne primaarsetes lihasprotsessides ja denervatsioonipotentsiaalide registreerimisel (fibrillatsioonide, fastsikulatsioonide ühe-, kahefaasilised potentsiaalid).
Globaalne (naha) EMG informatiivne neuraalsete ja seljaaju kahjustuste tasemete jaoks (II tüüpi EMG Jusevitši järgi), samuti perifeersete motoorsete neuronite funktsionaalse seisundi ning püramiid- ja ekstrapüramidaalsüsteemide struktuuride mõju uurimiseks neile.
ENMG võimaldab määrata perifeersete närvide aferentsete ja eferentsete kiudude impulsi kiirust. Lihaste esilekutsutud reaktsioonide ja närvi aktsioonipotentsiaali (AP) analüüs on informatiivne müelinopaatia (sensoriline ja/või neuromotoorne) ja aksonopaatia (sensoriinne ja/või neuromotoorne) diagnoosimisel. Stimulatsioon ENMG (rütmilise närvi stimulatsiooni meetod) tuvastab neuromuskulaarsed (sünaptilised) ülekandekahjustused.
Neurofüsioloogilised uuringud võimaldavad hinnata patoloogilise protsessi lokaliseerumist ja neuromotoorse aparaadi kaasatuse astet, samuti aitavad eristada erinevaid neuromuskulaarseid haigusi. Need meetodid on mitteinvasiivsed ja neid saab kasutada mitu korda.
Biokeemilised uuringud Need hõlmavad ensüümide, eelkõige kreatiinfosfokinaasi (CPK), laktaatdehüdrogenaasi (LDH) ja fruktoosdifosfaat-aldolaasi (FDA) aktiivsuse määramist, samuti kreatiin-kreatiniini indeksi muutusi. Nende ensüümide aktiivsus suureneb järsult progresseeruva lihasdüstroofia (PMD) korral, eriti protsessi varases staadiumis: CPK aktiivsus vereseerumis suureneb kümme korda, mõnikord 50 või enam korda; LDH aktiivsus - 5-7 korda; FDA tegevus - 2-5 korda. Hilisematel etappidel väheneb vereseerumi ensüümide aktiivsus normaalsetele väärtustele. Sekundaarsete neuraalsete ja spinaalsete amüotroofiate korral muutuvad ensüümide omadused suhteliselt vähe. CPK aktiivsus on väga tundlik lihaskahjustuse marker, kuid selle mõõdukat tõusu täheldatakse ka amüotroofse lateraalskleroosi korral pärast treeningut või krambihoogu.
Patoloogilised uuringud. PMD-ga patsientide skeletilihaste biopsia tulemuste kohaselt määratakse sidekoe proliferatsioon ning seljaaju ja neuraalsete amüotroofiate korral lihaskiudude denervatsiooniatroofia.
Neuromuskulaarsete haiguste ravi
NMD, sealhulgas PMD ravi on äärmiselt keeruline. Ravi raskused on seotud primaarse metaboolse defekti mõnikord võimatu kindlaksmääramisega teatud pärilike haiguste vormide korral, samuti nende haiguste, eriti primaarse PMD, pidevalt progresseeruva kuluga.
Ravi on suunatud haiguse arengu kiiruse pidurdamisele ja patsiendi eneseteenindusvõime maksimeerimisele. Ravi põhimõtted:
- skeletilihaste ainevahetuse korrigeerimine (ainevahetuse stimulandid, anaboolsed steroidid, kaaliumipreparaadid, vitamiinid);
- segmentaalse aparatuuri stimuleerimine (müostimulatsioon, neurostimulatsioon, biotagasiside – bioloogilised meetodid tagasisidet EMG, refleksoloogia, balneoteraapia,
Skeletilihaste põletikku nimetatakse müosiidiks. Seda haigust iseloomustab lokaalne valu, mida süvendab füüsiline pingutus, ja aja jooksul suureneb ebamugavustunde intensiivsus. Liikumised on järsult piiratud, ilmneb lihasnõrkus. Lihaspõletik areneb nakkuslike, viiruslike patoloogiate või mehaaniliste vigastuste tagajärjel.
Haigust võivad esile kutsuda nii sisemised kui ka välised põhjused. Endogeensete tegurite hulka kuuluvad:
Nakkushaiguste korral satub patogeen koos lümfi- ja verevooluga lihaskudedesse, põhjustades ägeda põletikulise protsessi. Nakkusliku etioloogiaga müosiidil on mädane ja mittemädane. Esimest täheldatakse inimestel gripi, tuberkuloosi, süüfilise ajal, kui nad on nakatunud Coxsackie viirusega (Bronholmi tõbi).
Mädane lihaspõletik areneb koos ulatusliku infektsiooniga organismis: streptokokk või stafülokokk, sepsis, luude osteomüeliit. Kudedes moodustuvad nekrootilised kolded, abstsessid, flegmoonid.
Põletiku eksogeensed põhjused on järgmised:
- lihaskrambid ujumise ajal;
- trauma;
- raske hüpotermia;
- krooniline lihaspinge;
- pikaajaline viibimine ebamugavas asendis.
Vigastuse ajal on kiud rebenenud, põhjustades ägedat põletikku, turset ja verejooksu. Pärast paranemist tekivad armid, lihas lüheneb, deformeerub, võivad tekkida luustumise piirkonnad.
Tänaval töötavatel inimestel võivad põletikuliseks muutuda selja-, emakakaela- ja nimmepiirkonna lihased. Müosiit kimbutab ka mehi ja naisi, kes on sunnitud pikka aega viibima ebamugavas asendis, näiteks muusikud, massöörid, autojuhid. Selle tulemusena on häiritud vereringe ja kudede toitumine, tekivad tihendid, järk-järgult arenevad düstroofsed protsessid.
Müosiidi klassifikatsioon
Vastavalt kahjustuse levimusele on müosiit lokaliseeritud ja generaliseerunud. Kohaliku tüübi korral muutub ainult üks lihasrühm põletikuliseks; polümüosiiti iseloomustab skeletilihaste mitme osa kahjustus korraga. Haigus võib areneda kaelas, alaseljas, puusades, säärelihastes, ribides, näol.
Sõltuvalt patogeneesist jaguneb polümüosiit järgmisteks tüüpideks:
- dermatomüosiit;
- neuromüosiit;
- luustuv müosiit;
- polüfibromüosiit.
Sõltuvalt kursuse kestusest on patoloogia äge ja krooniline, mis väljendub perioodiliste ägenemistena. Remissiooni staadiumis sümptomid taanduvad või kaovad täielikult.
Müosiidi lokaalse vormi sümptomid
Haiguse peamised sümptomid on äge valu lihastes palpatsiooni ja liikumise ajal. Ebamugavustunne suureneb ka öösel, kehaasendi muutumisega, muutusega kliimatingimused. Kuded on pidevalt pingeseisundis, mille tagajärjel on liigeste ja jäsemete liikuvus piiratud, inimene on sundasendis. Mõjutatud piirkonna nahk muutub kergelt punetavaks, puudutades soojaks.
Edaspidi areneb lihasnõrkus kuni osalise või täieliku atroofiani. Inimesel on raske tavalisi ülesandeid täita, ta kaotab eneseteenindusvõime. Progresseeruva kulgemise korral levivad sümptomid uutesse piirkondadesse. Näiteks rindkere, emakakaela piirkonna roietevaheliste lihaste põletik võib põhjustada kõri-, diafragma kahjustusi, köha, õhupuudust, peavalu, inimesel on raske neelata ja rääkida.
Algstaadiumis on märgatav lokaalne turse, punetus, nahaalused hemorraagid, kehatemperatuur tõuseb. Kui müosiidi põhjuseks on viirushaigused, ilmnevad lisaks üldise mürgistuse tunnused, külmavärinad, riniit, köha. 
Käe või jala lihaste põletikku diagnoositakse harva ja enamasti avaldub see üldistatult. Patsiendil on raske jäsemeid liigutada, sellega kaasneb tugev valu, lihasnõrkus. Inimene hoiab kätt või jalga kindlas mugavas asendis.
Kõige tavalisem haigustüüp on emakakaela müosiit. Samal ajal tekivad ebamugavad aistingud kuklas, kõrvades, abaluu all, migreenimured.
Mõnel juhul ei saa patsient oma kaela liigutada, õlalihased valutavad, närimisel tekib valu. Nimmepiirkonna müosiit mõjutab kudesid piki lumbosakraalse piirkonna selgroogu. Seda haigusvormi täheldatakse peamiselt eakatel inimestel.
Sõltuvalt haiguse staadiumist muutub lihaskoe konsistents. Esiteks muutuvad need tihedamaks, suurendavad mahtu, suurendatakse toonust. Järk-järgult lihased pehmenevad, moodustuvad sõlmed, luustumise piirkonnad. Deformatsioon põhjustab jäsemete erinevaid kontraktuure, kaela, selgroo kumerust.
Kuidas polümüosiit avaldub?
Polümüosiidi sümptomid ilmnevad kehas toimuvate autoimmuunprotsesside käigus. Kaitsesüsteem ebaõnnestub ja hakkab tootma tervetele rakkudele antikehi. Selle tulemusena toimub lihaskiudude hävitamine, mis viib põletikulise protsessini, mis levib naaberkudedesse ja organitesse. Seetõttu on polümüosiit sageli keeruline dermatiidi ja liigesekahjustuste tõttu.
Patoloogia üldistatud vormi diagnoositakse peamiselt keskealistel ja 5–15-aastastel lastel. Naised kannatavad lihaspõletike all palju sagedamini kui mehed. Polümüosiidi esimesteks sümptomiteks on nõrkus puusa-, õla- ja emakakaela piirkond. Tekivad raskused neelamisel, hingamisel, kõne aeglustub. Dermatomüosiidi korral ilmub naha pinnale lööve, lihased muutuvad järk-järgult õhemaks ja atroofeeruvad.
Polüfibromüosiiti iseloomustab asendamine lihaskoeühenduskohale. Kahjustatud rakkude asemel moodustuvad armid, sõlmed, adhesioonid. See toob kaasa kiudude lühenemise ja liikumisraskused, kuded on pidevas toonuses. Tihendid on palpatsioonil valusad, võivad perioodiliselt suureneda.
Neuromüosiidi korral hõlmab patoloogiline protsess närvilõpmed mis seda piirkonda innerveerivad. Patoloogia põhjustab tundlikkuse vähenemist või suurenemist, tuimust, kipitust, tugevat valu, toonuse langust, lihaspingeid, liigeste liikuvuse piiramist.
On iseloomulik, et tugevat valu piki närvitüvesid valuvaigistid ei peata, vaja on täiendavaid rahusteid.
Müosiidi ossificansi sümptomid arenevad polüfibromüosiidi taustal või pärast lihaskiudude rebenemist. Vigastuse kohas moodustunud sidekoe rakud immutatakse järk-järgult kaaliumi-, kaltsiumi- ja fosforhappesooladega. See viib teatud piirkonna luustumiseni. Järk-järgult kasvavad need kolded koos luudega, mis aitab kaasa jäsemete deformatsioonile.
Müosiidi luupõletiku kliinilised tunnused: lihaste kõvenemine, liikuvuse piiramine, teatud kehaosade deformatsioon, tugev valu treeningu ajal ja puhkeolekus. Kui käe või jala lihased ühinevad luudega, tekib jäseme täielik liikumatus.
Müosiidi diagnoosimise meetodid
Diagnoosi kindlakstegemiseks viib arst läbi patsiendi uuringu ja uuringu. Ravi ja uuringu määrab terapeut, neuroloog, reumatoloog või dermatoloog. Antikehade, T-lümfotsüütide kontsentratsiooni määramiseks on ette nähtud reumaatilised testid. Biokeemiline vereanalüüs näitab leukotsüütide, valgu sisaldust veres.
Vähkkasvajate välistamiseks hinnatakse kiudude kahjustusi biopsiaga. Selleks pigistage koetükk ära ja viige läbi tsütoloogiline, morfoloogiline uuring. Analüüs on ette nähtud müosiidi, polümüosiidi ja polüfibromüosiidi nakkusliku vormi jaoks. 
Erinevat tüüpi müosiidi ravi
Lihasvalu sündroomi leevendamiseks on ette nähtud kortikosteroidide, mittesteroidsete põletikuvastaste ravimitega salv. Kui müosiit ilmnes pärast kannatusi külmetushaigused või hüpotermia, viige läbi kohalik ravi geelidega: Dolobene, Apizartron, Indamethacin, Traumeel S.
Salvi hõõrutakse kahjustatud piirkonda 2-4 korda päevas. Ravimitel on valuvaigistav, ödeemi- ja põletikuvastane toime, nad parandavad verevoolu, vähendavad lihastoonust.
Polümüosiidi ravi viiakse läbi mittesteroidsete põletikuvastaste ravimite (Diklofenak, Indometatsiin), lihasrelaksantide (Mydocalm, Mefedol) süstidega. Peamine ravim autoimmuunse iseloomuga lihaste generaliseerunud põletiku ravis on prednisoloon. Sellel on põletikuvastane, immunosupressiivne ja allergiavastane toime. Prednisolooni manustatakse süstimise või suukaudse tabletina. Ambene kapslid vähendavad põletikku, omavad reumavastast toimet.
Kuidas ravida mädast müosiiti, millega kaasneb palavik ja mädakollete teke? Määratakse laia toimespektriga antibiootikumid, palavikuvastased ja valuvaigistid (Reopirin). Naha hõõrumine salvidega on vastunäidustatud, see võib suurendada põletikku.
Osteeruva müosiidi ravi toimub enamikul juhtudel kirurgiliselt. Ravim Hüdrokortisoon aeglustab patoloogilisi protsesse, takistab kaltsifikatsioonide ladestumist. Polüfibromüosiidi korral on kontraktuuride ja armide resorptsiooni vältimiseks ette nähtud mittesteroidsed põletikuvastased ravimid (Ibuprofeen), füsioteraapia, Lidaza süstid. Salv Gevkamen omab kohalikku ärritavat toimet, leevendab põletikku.
Lihaskudede põletikku võivad põhjustada nakkushaigused, vigastused ja autoimmuunprotsessid organismis. Patsient on mures terav valu kahjustuse korral jäsemete ja liigeste piiratud liikuvus. Ilma õigeaegse ravita on lihaste nõrkus, nende kiudude atroofia, kontraktuurid, sõlmed. Ravi määratakse, võttes arvesse haiguse staadiumi ja vormi.
Lihaspõletikud – haigused, mille peamisteks sümptomiteks on vöötlihaste põletikuga kaasnev lihasnõrkus. Lihaspõletik hõlmab idiopaatilisi põletikulisi müopaatiaid, infektsiooniga seotud müopaatiaid ning ravimite ja toksiinidega kokkupuutest tingitud müopaatiaid. Nende hulgas on kõige olulisemad polümüosiidi ja dermatomüosiidi nähud. Selles artiklis vaatleme lihaspõletiku sümptomeid ja peamisi lihaspõletiku tunnuseid inimestel. Lisaks räägime põletikuliste lihaste diagnoosimisest.
Lihase põletiku sümptomid
Lihaspõletiku sümptomite ilmnemisel märgib enamik patsiente halb enesetunne, üldine nõrkus, nahakahjustused (koos dermatomüosiidiga). Seejärel liituvad lihaste põletikuga järk-järgult (mitme nädala jooksul) proksimaalsete lihasrühmade nõrkuse progresseeruva suurenemise sümptomid. Mõnedel lihaspõletiku nähtudega patsientidel (lapsed ja noored) täheldatakse ägedat haigust, mis sageli kaasneb väljendunud põhiseaduslike tunnustega (palavik, kehakaalu langus jne) ja müalgiaga.
Väga aeglast (mitme aasta jooksul) lihasnõrkuse suurenemist koos lihaspõletiku sümptomitega täheldatakse sagedamini inklusioonmüosiidi all kannatavatel eakatel patsientidel. Äärmiselt harva tekib põletikuliste lihastega nn amüotroofne dermatomüosiit, mille peamiseks sümptomiks on väga pikka aega tüüpiline nahakahjustus. Antisüntetaasi sündroomiga patsientidel võivad lihase põletiku varajased nähud hõlmata Raynaud’ fenomeni, polüartralgiat või polüartriiti ja interstitsiaalsest kopsufibroosist tingitud hingeldust.
Lihasekahjustuse sümptomid põletiku ajal
Lihaspõletiku juhtiv kliiniline tunnus on üla- ja alajäsemete proksimaalsete lihasrühmade sümmeetriline nõrkus, samuti kaela paindumisel osalevad lihased. See muudab püsti tõusmise keeruliseks madal tool, maandumine transpordis, pesemisel ja kammimisel. Lihaspõletiku sümptomitega kõnnak muutub kohmakaks, kahlavaks, patsiendid ei saa ilma abita tõusta ja rebivad pead padjalt lahti. Neelu-, kõri- ja söögitoru lihaste põletik toob kaasa düsfoonia, neelamisraskused, köhahood. Distaalsete lihaste kahjustuse nähud on haruldased (10%), vähem väljendunud kui proksimaalsete lihaste kahjustus ja neid tuvastatakse peamiselt "lisadega" müosiidi korral. Pooltel patsientidest, kellel on lihaspõletiku sümptomid, lihasvalu või lihaste tundlikkus palpatsioonil, on võimalik lihaste turse, kuid lihaste atroofia areneb ainult pikka aega polümüosiidi/dermatomüosiidi all kannatavatel patsientidel, eriti adekvaatse ravi puudumisel. Lihashüpertroofia on lihasdüstroofiate iseloomulik tunnus ja seda ei täheldata polümüosiidi/dermatomüosiidi korral.
Nahakahjustuste sümptomid koos lihaste põletikuga
Dermatomüosiidi patognomooniline märk koos lihaste põletikuga. Nahanähtude hulka kuuluvad erütematoosne (heliotroopne) lööve, mis paikneb ülemistel silmalaugudel, põsesarnadel, ninatiibadel, nasolaabiaalse voldi piirkonnas, "dekoltee" piirkonnas ja ülaosas, küünarnukkide ja põlvede kohal, metakarpofalangeaalsed ja proksimaalsed interfalangeaalsed liigesed, pea peanaha osadel. Kergelt kõrgenenud või lamedaid erütematoosseid ketendavaid lööbeid, mis paiknevad sõrmenukkidel, nimetatakse "Gottroni märgiks" koos lihaspõletikuga. Iseloomulikud nahanähud, mida täheldatakse mitte ainult dermatomüosiidi, vaid ka polümüosiidi korral: peopesade naha punetus, koorumine ja lõhenemine ("mehaaniku või käsitöölise käsi"), küünenaha hüpertroofia, periungaalne erüteem, telangiektaasiad. Periunguaalse voodi lihaste põletikuga veresoonte kapillaroskoopia korral täheldatakse kapillaarsilmuste laienemist ja laienemist, sagedamini ristsündroomi, harvemini dermatomüosiidi korral. Fotodermatiit ja sügelus on vähem levinud.
Lihasepõletikuga liigesekahjustuse sümptomid
Liigesekahjustuse sümptomid eelnevad sageli arengule lihaste patoloogia lihaste põletikuga. Kõige sagedamini on haaratud käte väikesed liigesed, randme liigesed, harvemini - küünar- ja põlveliigesed. Kahjustus on kahepoolselt sümmeetriline, meenutades reumatoidartriidi oma, reeglina mööduv, glükokortikoidide määramisel kaovad lihaspõletiku sümptomid kiiresti. Küll aga on kirjeldatud kroonilise deformeeriva artriidi teket koos käte liigeste subluksatsioonidega, kuid ilma erosioonimuutusteta röntgenuuringu järgi.
Lupjumise sümptomid lihasepõletike korral

Lupjumise nähud ilmnevad hilisemates staadiumides, sagedamini juveniilse dermatomüosiidi korral. Lupjumised lokaliseeruvad subkutaanselt või sidekoes lihaskiudude ümber, sageli küünarnuki- ja põlveliigeste kohal asuvates mikrotraumapiirkondades, sõrmede ja tuharate painutuspindadel.
Kopsukahjustuse sümptomid koos lihaspõletikuga
Lihaspõletiku peamine kliiniline tunnus on väljahingamise hingeldus, mis võib olla seotud diafragmaalsete lihaste kahjustusega, südamepuudulikkuse tekkega, kaasuva kopsuinfektsiooni ja toksilise kopsukahjustusega, mis on seotud teatud ravimite, näiteks metotreksaadi võtmisega. Kirjeldatakse lihaspõletiku kliinilises pildis esile kerkiva ägeda difuusse alveoliidi sümptomite teket, mis väljendub ebaproduktiivse köha ja kiiresti progresseeruva hingamispuudulikkusena. Sagedamini täheldatakse interstitsiaalse kopsufibroosi aeglast progresseerumist, mis mõnel patsiendil tuvastatakse alles spetsiaalse läbivaatuse käigus. Kõige raskematel juhtudel areneb aspiratsioonipneumoonia.
Südamekahjustuse sümptomid koos lihaste põletikuga
Südamekahjustuse nähud polümüosiidi / dermatomüosiidi korral on enamikul juhtudel asümptomaatilised. Mõnikord ilmnevad spetsiaalse läbivaatuse käigus rütmi- ja juhtivushäirete sümptomid (tahhükardia, arütmia). Dilatatiivse kardiomüopaatiaga seotud südame paispuudulikkus on haruldane. Raynaud' fenomeni täheldatakse sagedamini dermatomüosiidi, antisüntetaasi sündroomi ja süsteemsete sidekoehaigustega polümüosiidi/dermatomüosiidi ristsündroomi korral.
Teiste märgid veresoonte häired lihaste põletikuga
Kirjeldatakse periunguaalse voodi, petehhiate, livedo reticularis'e (jäsemete ja kehatüve naha hargnenud muster) infarkte. Neerukahjustust täheldatakse harva, kuigi võib tekkida proteinuuria ja isegi nefrootiline sündroom. Raske müoglobinuuria võib põhjustada tsirroosi.
Lihase põletiku tunnused
Rakulised immuunvastused on polümüosiidi/dermatomüosiidi patogeneesis esmatähtsad. Mõjutatud lihase immunopatoloogiline uuring näitab aktiveeritud olekus T- ja B-lümfotsüütide ning makrofaagide infiltratsiooni. Samal ajal on T-rakkudel tsütotoksiline toime müofibrillide vastu. On märke teatud immunopatoloogilistest erinevustest polümüosiidi ja dermatomüosiidi vahel. Dermatomüosiidi korral on lihasinfiltraadis ülekaalus CD4+-T-lümfotsüüdid, makrofaagid ja B-lümfotsüüdid, polümüosiidi korral aga tsütotoksilised CD8+-T-lümfotsüüdid. Eeldatakse, et dermatomüosiidi tunnustega tekib humoraalne immuunvastus, mis viib komplemendi aktivatsioonini, mõjutades intramuskulaarseid mikroveresooneid ning polümüosiidi korral domineerivad tsütotoksilisi aineid (perforiin, gransüüm) sünteesivate CD8+-T-lümfotsüütide vahendatud rakulised tsütotoksilised reaktsioonid. Müosiidispetsiifiliste autoantikehade patogeneetiline tähtsus lihaste põletikul ei ole tõestatud.
Lihaspõletiku sümptomite põhjused
Lihaspõletike põhjused pole täpselt teada. Nakkuslike tegurite rollile viitab kaudselt ka sagedasem haigestumine talvel ja varakevadel (eriti juveniilse dermatomüosiidiga patsientidel), mis langeb ajaliselt kokku nakkuste epideemiatega. Geneetilise eelsoodumuse osalemist tõendab polümüosiidi / dermatomüosiidi tekke võimalus monosügootsetel kaksikutel ja patsientide veresugulastel. Mõnede suure h(HLA) antigeenide kandmine on tihedamalt seotud mitte lihaspõletiku endaga, vaid teatud immuunhäiretega, eelkõige müosiidispetsiifiliste autoantikehade hüperproduktsiooniga.
Lihaspõletiku nähtude levimus
Lihaspõletike esinemissagedus elanikkonnas on 2 kuni 10 juhtu 1 miljoni elaniku kohta aastas. Sõltuvalt vanusest täheldatakse kahte esinemissageduse tippu: 5-15-aastaselt (juveniilne dermatomüosiit) ja 40-60-aastaselt. Valdav sugu on naine (haigete naiste ja meeste arvu suhe on 2-3: 1)
Lihaspõletiku diagnoosimine
Lihase põletiku täielik vereanalüüs: iseloomulikud tunnused puuduvad, ESR-i suurenemist täheldatakse harva, peamiselt süsteemsete ilmingute tekkega.
Biokeemilised vereanalüüsid põletikuliste lihaste diagnoosimisel
Skeletilihaste kahjustuse üldtunnustatud mõõdik on CPK, mis suurendab polümüosiidi/dermatomüosiidi korral suurema tundlikkuse ja spetsiifilisusega kui teised laboriuuringud. CPK suurenemine koos lihaste põletikuga erinevad perioodid haigus esineb 95% polümüosiidi/dermatomüosiidiga patsientidest. CPK kontsentratsioon võib suureneda, kuni ilmnevad polümüosiidi/dermatomüosiidi ägenemise lihaspõletiku kliinilised nähud, ja selle tase võib langeda kuni kliinilise paranemiseni. Mõnikord võib patsientidel CPK tase olla normaalses vahemikus, hoolimata tõsistest lihaskahjustustest vastavalt morfoloogiline uuring, sel juhul ei ole indikaator korrelatsioonis aktiivsuse kliiniliste ja morfoloogiliste tunnuste dünaamikaga. Tuleb meeles pidada, et CPK normaalset taset võib täheldada raske lihasatroofiaga patsientidel haiguse hilisemates staadiumides, dermatomüosiidi debüüdi ja kasvaja müosiidi sümptomitega patsientidel.

Müokardi nekroosi puudumisel täheldatakse polümüosiidi/dermatomüosiidi nähtudega CPK MB fraktsiooni suurenemist. Transaminaaside aktiivsuse suurenemine ei ole skeletilihaste kahjustuste suhtes spetsiifiline. Mõnel üldise nõrkusega patsiendil tekitab isoleeritud transaminaaside tõus hepatiidi kahtlust.
Põletikuliste lihaste immunoloogiline diagnoos
Müosiidispetsiifilised AT-d hõlmavad ülekande-RNA aminoatsüülsüntetaaside AT-sid (antisüntetaasi AT-d), peamiselt histidüül-tRNA süntetaasi (Jo-1) AT-sid. AT Jo-1 tuvastatakse pooltel polümüosiidi/dermatomüosiidiga patsientidest, samas kui teised süntetaasivastased AT-d on äärmiselt haruldased (5%). Antisüntetaasi AT-de teket seostatakse nn antisüntetaasi sündroomi tekkega, mida iseloomustavad äge algus, interstitsiaalne kopsuhaigus, palavik, sümmeetriline artriit, Raynaud’ fenomen ja "mehaaniku käe" nahakahjustused lihaspõletiku ajal.
Instrumentaalsed meetodid lihaste põletiku määramiseks
Elektromüograafia lihaspõletike diagnoosimiseks on tundlik, kuid mittespetsiifiline meetod põletikuliste müopaatiate diagnoosimiseks. Tüüpilised sümptomid, mida täheldati rohkem kui 90% patsientidest proksimaalsete ja paraspinaalsete lihaste uurimisel, hõlmavad müofibrillide patoloogilise spontaanse aktiivsuse tunnuseid (fibrillatsioonipotentsiaalid, keerulised korduvad tühjenemised jne) stimulatsiooni ajal ja puhkeolekus, lühikesed madala amplituudiga mitmefaasilised potentsiaalid kokkutõmbumine. Tavaline elektriline aktiivsus elektromüograafia võimaldab enamikul juhtudel välistada polümüosiidi/dermatomüosiidi diagnoosi. Elektromüograafia on kasulik meetod lihaspõletike ravi efektiivsuse jälgimiseks, eriti kui laboratoorsete ja kliiniliste uuringute tulemused on küsitavad. Elektromüograafia andmed ei ole aga hästi korrelatsioonis lihasnõrkuse kliiniliste ilmingutega. On oluline, et steroidse müopaatia korral täheldataks samu (kuigi vähem väljendunud) muutusi kui aktiivse müosiidi korral.
Diagnoosi kinnitamiseks kasutatakse põletikunähtudega lihasbiopsiat isegi lihaspõletikule iseloomulike kliiniliste, laboratoorsete ja instrumentaalsete tunnuste esinemisel. Patoloogilises protsessis osaleva lihase kõige informatiivsem biopsia, kuid ilma tõsise atroofiata.
Röntgenuuringud lihaste põletiku diagnoosimiseks Liigespõletiku röntgensümptomid ei ole tüüpilised. Kopsude röntgenuuring näitab sageli basaalse pneumoskleroosi ja interstitsiaalse kopsufibroosi tunnuseid. Kõrge eraldusvõimega röntgen-CT (RCT) peetakse tundlikumaks meetodiks.
EKG lihaspõletiku nähtude diagnoosimisel. Prognoosiliselt ebasoodsate rütmi- ja juhtivushäirete varajaseks avastamiseks on soovitatav läbi viia igapäevane EKG monitooring (vastavalt Holterile).
Perifeersete närvide ja lihaste esmase kahjustusega degeneratiivsed haigused moodustavad olulise osa inimese pärilikust patoloogiast. Neuromuskulaarsete haiguste diagnoosimine põhineb molekulaargeneetilistel ja elektrofüsioloogilistel (EMG) uuringutel.
Elektroneuromüograafia võimaldab teil diagnoosi kinnitada ja jälgida haiguse dünaamikat. Neurogeense lihaspatoloogiaga saab tuvastada denervatsiooni tunnuseid: fibrillatsioonipotentsiaalid, positiivsed teravad lained, interferentsipotentsiaali amplituudi vähenemine, mitmefaasilised potentsiaalid. Primaarse lihase patoloogia korral on EMG pilt mittespetsiifiline ja muutuv; kõige iseloomulikum on potentsiaalide amplituudi vähenemine. Impulsside juhtivuse kiiruse (SPI) indikaatorid koos aksonopaatiaga on veidi vähenenud või on normi alumisel piiril. Demüeliniseerivate neuropaatiate korral väheneb SPI oluliselt. SPI ja aktsioonipotentsiaalide amplituudi muutmisega (tundlike või seganärvide poolt) saab diagnoosida tunnelneuropaatiat, samuti eristada aksonopaatiat ja müelinopaatiat. Hilinenud reaktsioonide varjatud perioodi pikenemist täheldatakse neuropaatiate ja radikulaarse sündroomi korral.
Diagnoosimisel mängivad olulist rolli biopsiaproovide uurimise morfoloogilised, immunohistokeemilised ja elektronmikroskoopilised meetodid. Lihaskiudude seisund valguse biomikroskoopias aitab eristada primaarset müogeenset atroofiat sekundaarsest denervatsioonist (neurogeenne või müelogeenne) amüotroofia. Biopsiaproovide histokeemiline analüüs on vajalik lihaskoe spetsiifiliste metaboolsete defektide tuvastamiseks. Elektronmikroskoopia on avanud terve klassi haigusi, mida ühendab mõiste "struktuurne müopaatia".
Ravi.Paljude lihaste, neuromuskulaarsete sünapside, perifeersete närvide ja motoorsete neuronite haiguste jaoks on välja töötatud etioloogiline ja patogeneetiline ravi. Muudel juhtudel on teraapia suunatud haiguse progresseerumise aeglustamisele, remissiooniperioodi pikendamisele ja patsiendi elukvaliteedi parandamisele. Neuromuskulaarsete haiguste ravi nõuab neuroloogide ja taastusravi spetsialistide ühist pingutust. Ravi taktika sõltub haiguse tõsidusest ja progresseerumise kiirusest.
Riis. 6.1.Pikaajalist hormoonravi saanud 13-aastase lapse välimus. Cushingoid
Pikaajalise kortikosteroidravi põhimõtted
Tüsistused sõltuvad annusest ja ravi kestusest (joonis 6.1). Peamised tüsistused: Cushingi sündroom, suhkurtõbi, osteoporoos, tuberkuloosi aktiveerumine, arteriaalne hüpertensioon, psühhoos, vastuvõtlikkus infektsioonidele, peptiline haavand.
Kortikosteroidide kaotamisega on võimalikud 3 tüüpi tüsistused. 1. Neerupealiste funktsiooni pärssimisega seotud tüsistused
cov. See areneb prednisolooni osalise tarbimisega annuses, mis ületab 20-30 mg päevas rohkem kui ühe nädala jooksul. Täielik taastumine võtab aega kuni aasta. Füsioloogilistele lähedastele annustele jääb neerupealiste funktsioon tavaliselt puutumatuks, kui ravi kestus ei ületa 1 kuud. Pärast kortikosteroidide tavaliste annuste manustamist ei ole asendusravi vajalik.
2. Üldised võõrutusnähud (anoreksia, iiveldus, oksendamine, unisus, peavalu, palavik, müalgia ja artralgia, kehakaalu langus) on tõenäolisemad pärast pikaajalist ravi. Ravi on sümptomaatiline, väikestes annustes kortisooni (10 mg päevas) mitme nädala jooksul.
3. Põhihaiguse ägenemine. See on üks kõige enam ohtlikud tüsistused pärast kortikosteroidide ärajätmist. Selle risk väheneb annuse järkjärgulise vähendamisega. Neuromuskulaarsete haiguste korral kasutatakse kõige sagedamini prednisolooni - suukaudseks manustamiseks mõeldud lühitoimelist ravimit. Seda võib manustada iga päev (jagatuna või üks kord hommikul) või ülepäeviti (üks kord hommikul). Lühikese kuuri (alla kuu) korral pole režiim hädavajalik. Pikaajalise ravi korral aitab osaline päevane tarbimine kaasa Cushingi sündroomi tekkele, pärsib neerupealiste funktsiooni ja vähendab vastupanuvõimet infektsioonidele. Pika kursusega singliga hommikune vastuvõtt lühitoimelise ravimi ööpäevane annus, mis vähem tõenäoliselt põhjustab
neerud (kuigi see ei takista Cushingi sündroomi esinemist). Ülepäeviti võtmisel tekib kahekordne ööpäevane annus vähem neerupealiste supressiooni, Cushingi sündroomi ja infektsioonide vastupanuvõime vähenemist. See skeem on efektiivne enamiku neuromuskulaarsete haiguste korral.
6.1. progresseeruvad lihasdüstroofiad
Termin "lihasdüstroofiad" viitab kliiniliselt polümorfsete geneetiliselt määratud haiguste rühmale, mis põhinevad esmastel progresseeruvatel degeneratiivsetel muutustel lihaskiududes. Müodüstroofia erinevad vormid erinevad üksteisest oma geneetilise olemuse, pärilikkuse tüübi, algusaja, lihaste atroofiate leviku topograafilise eripära poolest. Müodüstroofia iseloomulik kliiniline marker on "pardi" kõnnak, mis on seotud vaagnat reieluu suhtes fikseerivate tuharalihaste nõrkusega. Selle tulemusena toimub kõndimise ajal vaagna kalle mittetoetava jala suunas (Trendelenburgi fenomen) ja torso kompenseeriv kalle vastupidises suunas (Duchenne'i fenomen). Lisaks saavad patsiendid jälgida sõrmedel kõndimist, sagedasi kukkumisi, aeglast motoorset arengut ja spetsiifilisi piiranguid käte ülestõstmisel, trepist ronimisel, põrandalt tõusmisel.
Duchenne'i ja Beckeri müodüstroofia. Duchenne'i vorm on maailmas laialt levinud ja esineb sagedusega 1 3500 vastsündinud poisist, Beckeri vorm aga umbes 3-5 korda harvem.
Etioloogia ja patogenees. Duchenne'i ja Beckeri müodüstroofia on alleelsed variandid, pärilikud retsessiivsel X-seotud tüübil ja on põhjustatud kas sünteesi täielikust puudumisest või defektse suure molekulmassiga tsütoskeleti valgu-düstrofiini sünteesist. Düstrofiini puudumise tõttu kaotavad müofibrillid vastupanuvõime tsüklilistele kontraktsiooni-lõdvestumistele ja purunevad. Sarkoplasmaatilised membraanid muutuvad ebastabiilseks, ioonkanalite töö on häiritud, mille tulemusena suureneb vaba rakusisese ioniseeritud kaltsiumi kontsentratsioon, mis avaldab lihaskiude nekrotiseerivat toimet, põhjustades nende lüüsi (joon. 6.2).
kliiniline pilt. Esimesed kliinilised sümptomid ilmnevad enamikul Duchenne'i lihasdüstroofiaga poistel enne 3-5-aastaseks saamist: kõnnak on häiritud, lapsed hakkavad sageli kukkuma, kaotavad.

Riis. 6.2.Düstrofiini molekulaarne organisatsioon

Riis. 6.3.G. Duchenne'i kujutatud patsiendid
liikuvus. Säärelihaste pseudohüpertroofia tekkimine loob lihasjõust eksitava mulje (joonis 6.3). Pseudohüpertroofia võib areneda ka tuhara-, deltalihas-, kõhu- ja keelelihastes. Lõpuks muutub lihasnõrkus nii tugevaks, et laps ei tõuse peaaegu põrandalt, kõnnib "pardi" kõnnakuga, kasutab müopaatilisi võtteid: "ise ronimine", "redeliga ronimine" (Goversi sümptomid).
Riis. 6.4.1,5 aastane Duchenne'iga laps
Riis. 6.5.Sama laps 5-aastaselt. Lihaste pseudohüpertroofia, lordoos
Motoorsed funktsioonid stabiliseeruvad suhteliselt 3–6-aastaselt. Enamasti püsib võime kõndida ja trepist üles ronida kuni 8. eluaastani. 3–8-aastaselt toimub Achilleuse kõõluste edasine lühenemine ja hüppeliigeses moodustuvad fikseeritud fleksioonikontraktuurid, areneb kompenseeriv nimmepiirkonna hüperlordoos, lülisamba rindkere kyfoskolioos, reie-, vaagna- ja seejärel õlavöötme lihaste atroofia. , selg ja proksimaalsed käed. Tähelepanu juhitakse "lõdva õlavöötme", "pterügoidse abaluude", "herilase vöökoha" olemasolule. Sageli varjab lihaste atroofiat hästi arenenud nahaalune rasvakiht. Sageli tekivad rindkere ja jalgade deformatsioonid, hajus osteoporoos. Esmalt kaovad põlvekedra-, painutus- ja sirutajalihase küünarnuki refleksid, samas kui Achilleuse refleksid võivad püsida üsna pikka aega. 9-aastaselt liigub osa lapsi juba ratastooli abil, kuid enamusel iseseisva liikumise oskus kuni 12. eluaastani, püsti seismine 16. eluaastani. Hingamislihaste ja diafragma nõrkus põhjustab kopsude elutähtsuse vähenemise 20% -ni normist, mis põhjustab öise hüpoventilatsiooni episoode (joon. 6.4-6.6).
Mõnedel patsientidel ilmnevad erinevad endokrinopaatia tunnused: adiposogenitaalne sündroom, lühike kasv. Tähtaeg

Riis. 6.6.Sama poiss 14-aastaselt. Seljaaju väljendunud deformatsioon, paindekontraktuurid, lihaste atroofia

Riis. 6.7.Jalalihaste pseudohüpertroofia Beckeri tõve korral
düstrofiini - apodüstrofiinide - ajuisovormide puudulikkusega on mõnel Duchenne'i lihasdüstroofiaga patsiendil erineva raskusastmega vaimne alaareng. Laste vaimsete häirete raskusaste ei ole korrelatsioonis lihasdefekti raskusastmega ja müodüstroofse protsessi staadiumiga. Duchenne'i müodüstroofia kaugelearenenud staadiumi kohustuslik tunnus on hüpertroofiline ehk dilatatiivne kardiomüopaatia, millega kaasnevad südame rütmihäired, selle piiride laienemine ja südamepuudulikkuse sümptomid. Kardiomüopaatia on Duchenne'i müodüstroofia kõige levinum surmapõhjus. Hingamispuudulikkus, mis on provotseeritud vahelduvatest infektsioonidest või aspiratsioonist, põhjustab ka surma. Patsiendid surevad 2-3. elukümnendil.
Beckeri müodüstroofia (joon. 6.7) võib areneda pärast 15.
20 aastat, voolab palju pehmem. Selle müodüstroofia vormiga patsiendid jäävad ellu kuni täiskasvanueani. Intellektuaalne kahjustus ei ole talle iseloomulik, kõõluste tagasitõmbed ja kontraktsioonid on vähem väljendunud kui Duchenne'is, kardiomüopaatia võib puududa. Mõnel patsiendil tulevad aga südametegevuse häired esile ja on sageli haiguse sümptomiks. Lisaks säilib mõnel Beckeri müodüstroofiaga patsiendil viljakus, mistõttu täiskasvanud patsiendid võivad tütre kaudu haiguse edasi anda ka lapselastele ("vanaisa efekt").
Diagnostika.Duchenne'i müodüstroofiat iseloomustab ensüümide taseme märkimisväärne tõus juba müodüstroofia varases staadiumis.
füüsiline protsess. Alla 5-aastastel patsientidel võib kreatiinfosfokinaasi (CPK) tase ületada normi ülemist piiri kümneid ja isegi sadu kordi. Ensüümi kontsentratsioon väheneb seejärel ligikaudu 20% aastas. Samuti on kõrgenenud aldolaasi, laktaatdehüdrogenaasi ja transaminaaside tase seerumis. Kõrge CPK aktiivsus on praktiliselt haiguse kohustuslik tunnus ja lisaks Duchenne'i müodüstroofiale võib see tekkida ka Beckeri müodüstroofia (tavaliselt mitte üle 5000 U/l), polümüosiidi, dermatomüosiidi, hüpotüreoidismi, alkohoolse müopaatia ja paroksüsmaalse müoglobinuuriaga. EMG näitab primaarse lihase kahjustuse tunnuseid (madalpinge kõver koos mitmefaasiliste potentsiaalide rohkusega, motoorsete üksuste aktsioonipotentsiaalide lühenemine).
Praegu on Duchenne'i ja Beckeri müodüstroofia diagnoosimise, geenikandjate tuvastamise ja sünnieelse diagnoosimise üldtunnustatud "kuldstandardiks" mutatsioonianalüüs. Düstrofiini immunohistokeemilist reaktsiooni kasutatakse düstrofiini protsendi analüüsimiseks lihastes ja eristatakse Duchenne'i ja Beckeri vorme (algul see puudub). Heterosügootsetel kandjatel (patsientide emad ja õed) tuvastatakse ligikaudu 70% juhtudest skeletilihaste patoloogia subkliinilised tunnused: CPK suurenemine, esmased lihasmuutused EMG-l ja lihaste biopsia proovide uurimisel. Mõnikord on kandjatel säärelihaste paksenemine ja mahu suurenemine, suurenenud lihaste väsimus treeningu ajal, lihasspasmid pärast treeningut (kramp).
Luude röntgenuuring aitab tuvastada pikkade luude diafüüsi atroofiat, kortikaalse kihi hõrenemist, osteoartikulaarse kanali ahenemist, difuusset osteoporoosi.
Kardiovaskulaarsüsteemi kahjustus (kardiomüopaatia) areneb 73% haigetest lastest. Düstrofiini puudulikkus kardiomüotsüütides põhjustab kardiomüotsüütide progresseeruvat atroofiat ja nende asendamist kiudkoega. Kardiomüopaatia diagnoositakse esmakordselt 6-7-aastaselt, 20. eluaastaks on see 95% patsientidest. Samuti on tahhükardia, arütmia, pulsi ja vererõhu labiilsus, summutatud toonid, südame piiride laienemine. EKG näitab südame rütmihäireid, ventrikulaarseid ekstrasüstole, vasaku vatsakese hüpertroofia tunnuseid (27%): sügav hark K juhtmetes II-III aVF ja V 6 ; kõrge R pliis V 1, müokardi isheemia tunnused (5%). Echo-CG võib paljastada hüpertroofia (55%) või laienenud
(25%) kardiomüopaatia, kodade vaheseina defekt, mitraalklapi prolaps, vasaku vatsakese müksoom.
Südamelihase biopsia näitab lihaskiudude atroofiat, interstitsiaalset fibroosi, rasvade infiltratsiooni.
Duchenne'i ja Beckeri müodüstroofia diferentsiaaldiagnostika viiakse läbi kaasasündinud puusaliigese düsplaasia, D-vitamiini suhtes resistentse rahhiidi, proksimaalsete spinaalsete amüotroofiate, polümüosiidi ja dermatomüosiidi, metaboolsete ja endokriinsete müopaatiate korral.
Tüdrukute Duchenne'i müodüstroofia kliinilise fenotüübi esinemisel tuleks esmalt välistada X-autosomaalsete translokatsioonide või muude kromosoomide aberratsioonide esinemine X-kromosoomi huvides, aga ka mõned muud haruldased geneetilised variandid. Lisaks tuleb välistada Shereshevsky-Turneri sündroom (X-monosoomia). Sel eesmärgil viiakse läbi karüotüübi tsütogeneetiline uuring.
Emery-Dreyfuse müodüstroofia on aeglaselt progresseeruv müodüstroofia vorm, millel on X-seotud retsessiivne pärilikkus, mis on põhjustatud peamiselt skeleti-, silelihas- ja kardiomüotsüütides toodetava tsütoskeleti lihasvalgu - emeriini - geeni mutatsioonist.
Kliiniline pilt (joonis 6.8). Haigus algab 5–15-aastaselt. Varaseimad ja tüüpilisemad sümptomid on progresseeruvad paindekontraktuurid küünarliigestes ja käte sirutajalihastes, Achilleuse kõõluste tagasitõmbumine. Reeglina on 12-aastaselt patsientidel juba märkimisväärsed kontraktuurid põlve-, pahkluu- ja küünarliigeses. Siis on õla biitsepsi ja triitsepsi lihaste nõrkus ja atroofia, hiljem - deltalihas ja muud õlavöötme lihased. Mõnel juhul märgitakse esimese sümptomina varvastel ja jalgade välisservadel kõndimist, mis ilmneb umbes 5-aastaselt. Kuni selle hetkeni ei ole laste motoorne areng tavaliselt häiritud. Lihasnõrkus tekib märkamatult ja progresseerub aeglaselt. Umbes 20-aastaselt toimub suhteline stabiliseerumine. Säilib kõndimis- ja trepist ronimise oskus. Näolihaseid see ei mõjuta. Lihasnõrkus esineb kätes (õlavarreluu) ja jalgades (peroneaalne). Goveri manöövrid ja säärelihaste pseudohüpertroofia võivad puududa. Kõõluste reflekse ei kutsuta esile. Emakakaela tagumised lihased on sageli lühenenud, on piirang

Riis. 6.8.12-aastane Emery-Dreyfuse lihasdüstroofiaga patsient
lülisamba kaelaosa liigutused (lülisamba jäikuse sündroom). Haiguse sagedased ja prognostiliselt olulised sümptomid on südame juhtivuse häired ja arenev dilatatiivne või hüpertroofiline kardiomüopaatia. Kardiomüopaatiat võib komplitseerida siinussõlme südamestimulaatorite fibroosist tingitud kodade halvatuse teke. Sellistel juhtudel on näidustatud kunstliku südamestimulaatori kiire siirdamine.
Sünkoop ja bradükardia võivad mõnel juhul eelneda lihasnõrkuse tekkele, kuid enamasti esinevad need 3. elukümnendil. Südame juhtivussüsteemi muutusi ei tuvastata alati standardse EKG-uuringuga, kuid jälgimine võib paljastada atrioventrikulaarsed blokaadid ja Samoilov-Wenckebachi perioodid. Arütmia, mida ei saa kunstliku südamestimulaatori implanteerimisega korrigeerida, võib põhjustada insuldi ja patsiendi surma. Emery-Dreyfuse müodüstroofia elutähtis prognoos sõltub täielikult südamekahjustuse astmest.
Diagnostika.CPK aktiivsus suureneb mõõdukalt, laktaatdehüdrogenaasi ja aldolaasi aktiivsus - vähemal määral. Emery-Dreyfuse lihasdüstroofia kasuks annab tunnistust immunofluorestsentsreaktsiooni puudumine 12 monoklonaalse antikehaga emeriini suhtes leukotsüütide, lihaste ja naha biopsiate biomikroskoopias. Seda haigust iseloomustavad primaarsete lihaste ja neurogeensete kahjustuste kombineeritud EMG tunnused, millel on suur spontaanse denervatsiooni aktiivsus.
Näo-õla-õla müodüstroofia (Landuzi-Dejerine tüüp). Haigus on päritud autosoomselt domineerival viisil, millel on kõrge penetrantsus ja varieeruv ekspressiivsus. Seda esineb sagedusega 2,9 100 000 elaniku kohta. Tehti kindlaks näo-õla-õla müodüstroofia geneetiline heterogeensus. Enamik juhtumeid on seotud 4. kromosoomi pika käe mutatsiooniga.
kliiniline pilt. Haigus algab tavaliselt 2. elukümnendil. Esialgu täheldatakse atroofiat õlavöötmes, hiljem levib see näole. Patsientidel on näoilmed ammendatud; kõne muutub segaseks. Haiguse kõrgpunktis paiknevad suu ja silmade ringikujulised lihased, suur rinnalihas, eesmine serratus ja trapetslihase alumised osad, selja- latissimus dorsi, biitseps ja triitsepsõlg. Iseloomulikud sümptomid on "põiki naeratuse" ("Gioconda naeratus"), ülahuule väljaulatuvuse ("tapir huuled") kujul. Rind on lamestatud anteroposterioorses suunas, õlaliigesed on pööratud sissepoole, abaluud omandavad pterigoidse kuju. Atroofiad levivad allapoole. Kui protsessi on kaasatud jalalihased, on nõrkus kõige märgatavam peroneaalses lihasrühmas - "rippuvas jalas". Iseloomulik on asümmeetriline atroofia. Võib täheldada lihaste pseudohüpertroofiat. Kõõluste kontraktsioonid ja tagasitõmbed on mõõdukalt väljendunud. Kardiomüopaatia on haruldane. Võrkkesta veresoonte anomaaliaid angioretinograafias peetakse haiguse üheks fenotüübiliseks ilminguks. Silma raskete sümptomitega kaasneb telangiektaasia, turse ja võrkkesta irdumine. Võib tekkida kuulmislangus. Telangiektaasiad elimineeritakse koagulatsiooni teel, mis takistab pimeduse teket. Haiguse kulg on suhteliselt soodne. Füüsiline ülekoormus, intensiivne sportlikud tegevused ja irratsionaalne füsioteraapia võib aidata kaasa haiguse raskemale kulgemisele. Paljud haiged
püsivad funktsionaalselt ja nende elukvaliteet ei halvene. Teised seda haigust põdevad patsiendid on täiskasvanueas ratastoolis.
Diagnostika.CPK tase võib tõusta 5 korda. EMG registreerib nii müopaatilised motoorsed üksused kui ka denervatsioonipotentsiaalid. Paljudes jäsemete lihastes on histoloogilised muutused minimaalsed; abaluuülestes lihastes leitakse progresseeruv degeneratsioon ja marginaalne denervatsioon. On vaja välistada myasthenia gravis ja ajutüve kasvaja.
Jäsemete vöö müodüstroofia (CPMD) - proksimaalse lihasnõrkuse juhud, mis hakkab arenema 2. või 3. elukümnendil, progresseerub aeglaselt ja viib sügava puudeni alles 15-20 aasta pärast.
Etioloogia ja patogenees. CMDD ei ole geneetiliselt homogeenne; Praeguseks on tuvastatud umbes 10 erinevat geneetilist defekti.
kliiniline pilt. Esimesena kannatavad õla- ja vaagnavöötme lihased. Kaugelearenenud staadiumides on selja- ja kõhulihased märkimisväärselt mõjutatud, moodustub nimmepiirkonna hüperlordoos. Näo lihaseid tavaliselt ei mõjutata. Patsiendid näitavad tüüpilist "pardi" kõnnakut, müopaatilisi võtteid. Lihaste kontraktsioonid ja pseudohüpertroofia ei ole iseloomulikud. Kardiomüopaatia ei arene; intelligentsus säilib. Mehed ja naised on võrdselt mõjutatud. Surm võib tekkida kopsutüsistuste tõttu.
Diagnostika.CPK sisaldus on mõõdukalt suurenenud. EMG-l on esmase lihase kahjustuse tunnused. CMMD-d tuleb eristada Beckeri müopaatiast, juveniilsest seljaaju amüotroofiast, glükogeeni säilitamise müopaatiast, endokriinsetest, toksilistest, ravimitest põhjustatud müopaatiast, polümüosiidist ja müosiidist.
6.2. Kaasasündinud struktuursed müopaatiad
Kaasasündinud struktuursed müopaatiad (SCM) on aeglaselt progresseeruvate skeletilihaste haiguste geneetiliselt heterogeenne rühm. Erinevate SCM-i kliinilised sümptomid on mittespetsiifilised. Peamine kliiniline sümptom on hajus lihaste hüpotensioon, mis võib siiski esineda emakas ja määrata haruldase loote liikumise. SCM kuulub märkimisväärse osa nn loid lapse sündroomi põhjuste hulgas. Hüpotensioon valitseb vaagnavöötme lihastes ja pro-
jalgade sarnased osad. Vähemal määral on mõjutatud õlavöötme ja käte lihased. Sageli tuvastatakse puusa kaasasündinud nihestus, dolichocephalic pea kuju, gooti suulae, hobusejalg, kyphoscoliosis, lihaste hüpoplaasia. Iseloomulik on motoorse arengu hilinemine: lapsed hakkavad pead tõstma, istuma, tõusma, hilja kõndima, kõndides sageli kukkuma ega suuda joosta. Edaspidi ei saa teha lihtsamaid võimlemisharjutusi, osaleda õuemängudes. Patsientide kõõluste refleksid võivad olla normaalsed, vähenenud või puududa. SCM-i äärmiselt oluline kriteerium on progresseerumise puudumine või lihasnõrkuse väga aeglane suurenemine. Mõnel juhul võivad motoorsed funktsioonid vanusega mõnevõrra paraneda.
Diagnostika.CPK aktiivsus on normaalne või veidi suurenenud. EMG registreerib motoorsete üksuste madala amplituudiga polüfaasilisi müopaatilisi potentsiaale. Impulsi juhtivuse kiirus mööda motoorseid ja sensoorseid kiude on normaalne. Diagnoos on usaldusväärselt kindlaks tehtud ainult lihase biopsia tegemisel valgus- ja elektronmikroskoopia abil, mis paljastab lihaskiu spetsiifilise struktuuri. Haigete laste lihaste biopsiaproovide uurimine võib paljastada unikaalseid histoloogilisi tunnuseid, mis on määranud mitmeid nimetusi: tsentraalse varda haigus, müotubulaarne müopaatia, mittekarmiinne müopaatia, kolmekihiline müopaatia, I tüüpi kiudude lüüsi müopaatia, kerakeha müopaatia, müopaatia kehade kuhjumine "sõrmejälgede" kujul, müopaatia tsütoplasmaatiliste inklusioonidega vähenenud kehade kujul, müopaatia tuubulite agregatsiooniga jne.
Lihasdüstroofia ravi. Müodüstroofia ravivõimalused on oluliselt piiratud. Etioloogilist ja patogeneetilist ravi praktiliselt ei eksisteeri. Sümptomaatiline ravi on suunatud olemasoleva lihasjõu säilitamisele nii kaua kui võimalik, atroofia vähendamisele ja kontraktuuride tekke vältimisele. Peamine ülesanne on pikendada tegevusperioodi maksimaalse võimaliku perioodi võrra.
Kompleksne ravi koosneb medikamentoossest ravist, füsioteraapiast, ravivõimlemisest ja massaažist, ortopeedilisest korrektsioonist ja dieedist. Olulist rolli mängib psühholoogiline tugi, täiendõpe ja õige erialane orientatsioon.
Füsioterapeutilised protseduurid hõlmavad prozeriini, kaltsiumkloriidi, sinusoidselt moduleeritud või diadünaamiliste erinevate läbitungimisvõimetega voolude elektroforeesi, elektromüostimulatsiooni, osokeriidi, parafiini ja muda aplikatsiooni, vannid (radoon, okaspuu, väävel, vesiniksulfiid). Soovitatav on oksübaroteraapia, kuna hapnik pärsib fibroosi ja kollageeni moodustumist. Konservatiivse (spetsiaalsed lahased ja stiil) ja operatiivse iseloomuga (achilleotoomia, müotoomia) ortopeediline korrektsioon on suunatud kontraktuuride ja tekkivate patoloogiliste jäsemete vastu võitlemisele ning samuti on eesmärgiks säilitada patsiendi võime iseseisvalt liikuda. Igal juhul on vaja individuaalselt kaaluda operatsiooni eeldatavat kasu ja võimalikku kahju. Termiliste protseduuride järgselt tekkivate kontraktuuride korral on soovitatav lihaseid hoolikalt venitada kuni 20-30 korda päevas, millele järgneb une ajal lahastamine.
Patsiendile soovitatakse optimaalse ja tasakaalustatud vitamiinide ja mikroelementide sisaldusega proteiinisisaldusega, rasvade (eriti loomse päritoluga) ja süsivesikute piiranguga dieeti. Vältida tuleb soolaseid, praetud, maitseaineid, marinaade, kangeid lihapuljongisid, kohvi, šokolaadi, kakaod, kooke, küpsetisi.
Narkoteraapia eesmärk on kompenseerida lihaskoe energiapuudust, parandada kudede ainevahetust ja vereringet ning stabiliseerida lihaskiudude membraane. Kandke nikotiinhapet, vitamiine B 6, B 12, A ja E (aevit). Valkude sünteesiprotsesside parandamiseks kasutatakse aminohappepreparaate (tserebrolüsiin, glütsiin, metioniin, glutamiin, foolhapped). Välja on kirjutatud mittesteroidsed anaboolsed ained (kaaliumorotaat), makroergilised ravimid (fosfaden), kardiotroofsed ained (riboksiin, karnitiinkloriid, solkoserüül), perifeerset vereringet parandavad ained (trentaal, halidor, teonikool, oksübraal) ja nootroopsed ained (pantogaam, piratsetaam)] (nootropiil) . Mitokondriaalses hingamisahela süsteemis toimuvate energiaprotsesside parandamiseks kasutatakse koensüümi Q10 (ubikinooni), limantari, tsütokroom-C intravenoosseid infusioone. Võõrutus ja vere reoloogiliste omaduste parandamine, slaidi sündroomi leevendamine saavutatakse vasoaktiivsete ravimite infusioonide, reopolüglütsiini ja plasmafereesikursuste abil. Rakumembraanide suhtelist stabiliseerumist soodustavad prednisolooni väikesed annused. Paranduseks
kardiomüopaatia kasutada kardiotroofseid ravimeid (välja arvatud hüpertroofilise kardiomüopaatiaga patsiendid); südamepuudulikkuse korral - südameglükosiidid, diureetikumid, kaptopriil. Südame rütmihäiretega on ette nähtud kinidiin, β-blokaatorid, kaltsiumi antagonistid. Täieliku atrioventrikulaarse blokaadi väljatöötamisega muutub aktuaalseks küsimus kunstliku südamestimulaatori implanteerimise otstarbekuse kohta.
Mõnede müodüstroofiate (Duchenne'i ja Beckeri haigused) geeniteraapia meetodite väljatöötamise väljavaated on seotud geenitehnoloogiate täiustamisega. Aktiivne otsitakse geneetilisi kandjaid (vektoreid), mis suudavad sisestada düstrofiini geeni või minigeene haige retsipiendi lihasrakkudesse. Erakordselt tähtsaks peetakse perekonna meditsiinilist geneetilist nõustamist, sünnieelset diagnostikat koos loote DNA uurimisega.
6.3. Lülisamba lihaste amüotroofiad
Lülisamba lihaste amüotroofiad (SMA) on perifeerse närvisüsteemi pärilike häirete heterogeenne rühm. Patogenees on seotud seljaaju eesmiste sarvede (mõnel juhul ajutüve motoorsete tuumade) motoorsete neuronite progresseeruva degeneratsiooniga. Selle põhjuseks on programmeeritud rakusurma põhjustav geneetiline defekt – raku apoptoos. Motoorsete neuronite kadumine põhjustab vöötlihaste lõtva halvatuse ja denervatsiooni atroofiat. Enamasti esineb jäsemete proksimaalsete lihaste sümmeetriline kahjustus; distaalne amüotroofia, pirni kahjustus
harvemini arenevad kangilihased ja kahjustuse asümmeetria. Keskne motoorne neuron on tavaliselt terve. Sensoorsed häired puuduvad.
SMA erinevad variandid erinevad alguse vanuse, kulgemise olemuse, skeletilihaste kahjustuse topograafia ja pärilikkuse tüübi poolest (joon. 6.9). Enamik vorme päritakse autosoomselt retsessiivsel viisil. Iseloomustab mitmeid vorme

Riis. 6.9.Lõdva beebi sündroom SMA-s
autosomaalsed domineerivad ja X-seotud retsessiivsed pärimismustrid. Lihasbiopsia histoloogiline uurimine näitab, et väikese suurusega lihaskiud, hüpertroofiliste ja atroofiliste lihaskiudude kimbud külgnevad normaalse suurusega kiudude rühmadega.
Kui EMG näitab vaieldamatuid SMA sümptomeid, ei ole lihaste biopsia vajalik. SMA ravi ja taastusravi põhimõtted on samad, mis müodüstroofia puhul. Etiotroopset ja patogeneetilist ravi pole veel välja töötatud.
Proksimaalsed spinaalsed amüotroofiad lapsepõlves on päritud autosoomselt retsessiivselt. Fenotüüpseid on kolm erinevaid valikuid, mis erinevad kliiniliste ilmingute vanuse, kulgemise ja prognoosi poolest:
I tüüpi ehk Werdnig-Hoffmanni äge pahaloomuline infantiilne SMA;
II tüüp ehk krooniline infantiilne SMA (keskmine tüüp);
III tüüp ehk alaealine Kugelberg-Welander SMA.
Need põhinevad ühel geneetilisel mutatsioonil – geeni deletsioonil, mis on vajalik motoorse neuroni elujõulisuse tagamiseks. pikk õlg kromosoom 5. Mutatsioonide otsimine toimub DNA diagnostika käigus, sh lootel sünnieelse diagnoosimise käigus, mis aitab vältida haige lapse sündi.
Äge pahaloomuline infantiilne seljaaju amüotroofia (Werdnig-Hoffmanni tõbi või I tüüpi SMA) esineb sagedusega 1 25 500 vastsündinu kohta. Kliinilised sümptomid ilmnevad juba sündides või ilmnevad enne 6 elukuud. Veel emakas täheldatakse aeglast segamist, mis viitab loote motoorse aktiivsuse vähenemisele. Haigel lapsel on üldine nõrkus, peamiselt proksimaalses piirkonnas lihasrühmad ah, hüpotensioon ja arefleksia. Seljaasendis täheldatakse puusade paljunemise ja välise pöörlemisega "konnapoosit". Näolihased on suhteliselt terved, okulomotoorsed lihased ei osale. Hingamisteede funktsioon on esialgu piisav. Avastatakse keele atroofia ja fastsikulatsioonid, käte fascikulaarne treemor. Bulbar-sündroomi väljakujunemisel kaob neelu refleks, toitmine muutub palju raskemaks, mis võib põhjustada aspiratsioonikopsupõletikku. Tihti moodustub rindkere deformatsioon (joon. 6.10). Kui lihasnõrkus

Riis. 6.10.Laps, 6 kuud vana, Werdnig-Hoffmanni tõvega
avastatakse kohe pärast sündi, siis surm saabub umbes 6 kuu vanuselt, kusjuures esimeste sümptomite ilmnemisel 3 elukuu pärast võib elulemus olla umbes 2 aastat. Peamine surmapõhjus on hingamispuudulikkus kaasuvate hingamisteede haiguste taustal (joon. 6.11, 6.12).
Diagnoosimiseks tuvastatakse geenimutatsioon molekulaargeneetilise analüüsiga. CPK kontsentratsioon on tavaliselt normaalne, kuid kiiresti progresseeruva nõrkusega lastel võib see veidi tõusta. EMG tuvastab virvendus- ja fascikulatsioonipotentsiaalid puhkeolekus ning motoorsete üksuste potentsiaalide keskmise amplituudi tõusu. Juhtivuse kiirus mööda perifeersete närvide motoorseid aksoneid vastab reeglina normile. I tüüpi SMA-d tuleb eristada teistest haigusseisunditest, mis põhjustavad lõtva beebi sündroomi. Nende hulka kuuluvad kaasasündinud müodüstroofia ja neuropaatia, struktuursed müopaatiad, kaasasündinud või vastsündinu müasteenia gravis, metaboolsed müopaatiad, emakasisene poliomüeliit, botulism, kromosomaalne patoloogia, tserebraalparalüüsi atooniline vorm, Marfani sündroom.
Krooniline infantiilne seljaaju amüotroofia (II tüüpi SMA). Lihasnõrkus ilmneb tavaliselt 6. ja 24. elukuu vahel. Mida varem sümptomid ilmnevad, seda pahaloomulisem on kulg. Esialgsed nõrkuse ilmingud on tavaliselt sümmeetrilised ja neid täheldatakse jäsemete proksimaalsetes lihasrühmades. Reielihaste nõrkus on kõige märgatavam sümptom. Varasel perioodil on distaalne lihasnõrkus minimaalne või puudub üldse. Mõjutatud lihaste kõõluste refleksid vähenevad järsult. Kõik patsiendid on võimelised istuma, enamik suudab iseseisvalt seista ja mõned saavad isegi kõndida (joonis 6.13). Miimilised lihased

Riis. 6.11.Poiss, 5 aastat vana, Werdnig-Hoffmanni tõvega

Riis. 6.12.Poiss, 3 aastat vana, Werdnig-Hoffmanni tõvega

Riis. 6.13.Tüdruk, 9-aastane, Kugelberg-Welanderi tõvega
ja silma välised lihased haiguse algstaadiumis ei ole kahjustatud. Lihasnõrkus areneb aeglaselt. Mõnel juhul püsib see mitu aastat stabiilsena ja seejärel jätkub progresseerumine. Eeldatakse, et patsiendid elavad kuni täiskasvanueani, kuid isegi suhtelise stabiliseerumise perioodil, näitab EMG
fibrillatsiooni ja fascikulatsiooni potentsiaal. Moodustunud kontraktuurid, jalgade equinovarus deformatsioon. Juba imikueas täheldatakse lastel selgroo kõverust, rindkere deformatsioone, puusaliigeste düsplaasiat.
Diagnostika.CPK kontsentratsioon on normaalne. tulemused geneetiline analüüs ja EMG andmed on samad, mis ägeda infantiilse vormi puhul.
Juveniilne seljaaju amüotroofia (Kugelberg-Welanderi tõbi või III tüüpi SMA) esineb üldpopulatsioonis sagedusega 1,2 100 000. Motoorse aktiivsuse emakasisesel perioodil on piisav; laps on sündides terve. Sümptomid ilmnevad 2. ja 15. eluaasta vahel. Lapsed hakkavad ebakindlalt kõndima jalgade proksimaalse lihasnõrkuse tõttu. Gastrocnemius lihaste pseudohüpertroofia areneb, mis sageli põhjustab Duchenne'i lihasdüstroofia vale diagnoosi. Haigus kulgeb healoomuliselt, areneb väga aeglaselt. Harjad on mõjutatud hiljem. Näolihased võivad olla nõrgenenud, kuid liigutused silmamunad alati täies mahus. Bulbari häired ei ole iseloomulikud. Ligikaudu pooltel patsientidest võivad tekkida luude deformatsioonid, aeg-ajalt kõõluste tagasitõmbed ja kontraktuurid liigestes. Nõrgenenud lihaste kõõluste refleksid puuduvad või on oluliselt alla surutud. Sageli registreeritakse käte fascicular treemor.
Diagnostika.Ülimalt tähtis on geneetilise mutatsiooni tuvastamine. CPK kontsentratsioon võib ületada normi ülemist piiri 2-4 korda. Pooled EMG-ga patsientidest registreerisid spontaanset aktiivsust (fatsikulatsioonid, fibrillatsioonid ja positiivsed teravad lained). Lihaspinge korral täheldatakse amplituudi ja polüfaasi suurenemist, kestuse pikenemist ja motoorsete üksuste potentsiaalide arvu vähenemist. Juhtivus piki närvide tundlikke kiude on alati normaalne. Pika haiguse kulgemise korral võib juhtivuse kiirus mööda motoorseid kiude väheneda. III tüüpi SMA eristub jäsemete vöö müodüstroofiast.
Kennedy bulbospinaalne amüotroofia - SMA haruldane X-seotud retsessiivne vorm, mis debüteeris 4. elukümnendil; aeg-ajalt ilmnevad sümptomid 12–15-aastaselt. Geen on kaardistatud X-kromosoomi pikal käel. Mutatsioon mõjutab androgeeni retseptori geeni, sealhulgas spinaalseid motoorseid neuroneid, mis neid tekitab
retseptorid, mis pole tundlikud meessuguhormoonide (androgeenide) toime suhtes. Kliinilise pildi tuumaks on nõrkus, atroofia ja fastsikulatsioonid jäsemete proksimaalsetes lihasrühmades, kõõluste arefleksia, näo nõrkus, atroofia ja sidekulatsioonid keeles, perioraalsed fastsikulatsioonid, düsartria ja düsfaagia, treemor ja valulikud lihasspasmid (krambid). Harva areneb aksonaalne neuropaatia. Bulbaarsed häired tekivad tavaliselt 10 aastat pärast haiguse algust. Iseloomulikud on endokriinsed häired: günekomastia, munandite atroofia, potentsi ja libiido langus, suhkurtõbi, asoospermiast tingitud viljatus. Haiguse prognoos on üldiselt soodne: säilib kõndimisvõime ja enesehoolduse võimalus. Oodatav eluiga ei vähene, küll aga suureneb risk hormonaalsest tasakaalutusest tingitud pahaloomuliste kasvajate tekkeks (sh rinnavähk).
Diagnostika.Praegu on võimalik teha otsest DNA diagnostikat, tuvastada heterosügootset kandumist ja teha sünnieelset diagnostikat. EMG näitab denervatsiooni märke. CPK tase võib olla normaalne. Haigust tuleb eristada amüotroofsest lateraalskleroosist.
6.4. Mitmekordne kaasasündinud artrogrüpoos
Mitmekordne kaasasündinud artrogrüpoos on sündroom, mille peamiseks ilminguks on liigeste liikuvuse piiramine koos nende deformatsioonidega. Tavaliselt on kahjustatud distaalsed liigesed (pahkluu, randme), harvem - põlve- ja küünarliigesed. Lihasnõrkus artrogrüpoosi korral võib oma olemuselt olla nii neurogeenne kui ka müogeenne. Valdav enamus juhtudest on juhuslikud, ülejäänud juhtumid on päritud autosomaalselt retsessiivselt või X-seotud viisil. Neurogeense artrogrüroosiga täheldatakse haiguse kõige aktiivsemat faasi sünnieelsel perioodil ning juba vastsündinu perioodil on hingamine ja neelamine häiritud; mõned lapsed surevad aspiratsiooni tõttu. Kergematel juhtudel on ellujäämine parem ja lihasnõrkus progresseerub väga aeglaselt või ei edene üldse. Hiljem kaovad hingamishäired ja toitumisprobleemid. Kontraktuurid esinevad nii proksimaalsetes kui ka distaalsetes liigestes. Mõnel vastsündinul on seostatud mikrognaatiat, kõrge suulae ja näo kõrvalekaldeid, nagu näiteks
Edwardsi sündroom (trisoomia 18). Mõnedel neurogeense artrogrüroosiga lastel on eesaju arengus kõrvalekaldeid. On kombinatsioone meningomüelotseeli, mikrotsefaalia ja vaimse alaarenguga. Müogeense artrogrüpoosi sündroomi võib täheldada müopaatia korral koos kiutüüpide ebaproportsionaalsusega, kaasasündinud müodüstroofia, müotoonilise düstroofia, müasteeniliste sündroomide, fosfofruktokinaasi puudulikkusega.
Diagnostika.Lihaste histoloogilisel uurimisel ilmnevad iseloomulikud denervatsiooni ja reinnervatsiooni tunnused. Samuti ilmnevad müopaatia ilmingud: kollageenikiudude ja rasvkoe osakaalu suurenemine, keskmise suurusega kiudude kaootiline paigutus, lihaste spindlikapslite fibroos.
6.5. Põletikulised müopaatiad
Dermatomüosiit on süsteemne immuunsõltuv angiopaatia, mille puhul täheldatakse veresoonte ummistusi ja infarkte, mis viivad kõigi iseloomulike patoloogiliste muutuste tekkeni lihastes, sidekoes, nahas, seedetraktis ja närvikiududes. Patogenees on seotud antikehade ja immuunkomplekside moodustumisega ning komplemendisüsteemi aktiveerumisega. Perivaskulaarse infiltraadi koostis sisaldab T-lümfotsüüte, mis on valdavas enamuses T-abistajad, B-lümfotsüüdid ja plasmarakud.
kliiniline pilt. Esinemissageduse tipp saabub 5-10 aasta vanuselt, kuid juhtumeid üle varajane algus(kuni 4 kuu vanuselt). Sümptomid ilmnevad järk-järgult või välkkiirelt. Varjatud algust iseloomustavad palavik, halb enesetunne ja isutus (anoreksia). Lihasnõrkus võib sel ajal puududa. Need mittespetsiifilised sümptomid püsivad nädalaid kuni kuid, mis viitab püsivale infektsioonile. Enamikul lastel ilmneb dermatiit enne müosiiti. Lööve paikneb esialgu ülemistel silmalaugudel ja näeb välja nagu
erüteem koos kahjustatud pigmentatsiooni ja tursega. Seejärel levib see silmade ümber ja põskede piirkonda. Interfalangeaalsete, küünarnuki- ja põlveliigeste sirutajapindade erüteem ja tursed tekivad hiljem. Aja jooksul muutub nahk atroofiliseks ja ketendavaks. Müopaatilised muutused hõlmavad proksimaalset nõrkust, lihaste jäikust ja valu. Nõrkus suureneb, paindekontraktuurid ja liigeste deformatsioonid arenevad kiiresti. Kõõluste refleksid vähenevad ja seejärel kaovad. 60% patsientidest leitakse kaltsifikatsioone nahaaluskoes, eriti nende nahapiirkondade all, kus pigmentatsioon on häiritud. Mitmed lupjumised tekitavad röntgenikiirgusele "soomuse" efekti. Mõnel lapsel on esialgne sümptom lihaste jäikus ning naha- ja müopaatilised sümptomid on vähem väljendunud. Seedetrakti infarktid haiguse lõppstaadiumis on minevikus põhjustanud surma. Suremus dermatomüosiidisse on praeguseks vähenenud ja jääb alla 5%, mis on seotud ravimeetodite paranemisega. Rohkem kui 30% dermatomüosiidiga täiskasvanutest diagnoositakse hiljem pahaloomuline kasvaja.
Diagnostika.Palaviku, lööbe, müalgia ja nõrkuse kombinatsioon toetab dermatomüosiidi diagnoosimist. Haiguse alguses on CPK tase tavaliselt kõrgenenud. Aktiivse dermatomüosiidi ajal näitab puhkeoleku EMG virvendusi ja positiivseid teravaid laineid; juures lihaspingeid registreeritakse lühendatud madala amplituudiga mitmefaasilised potentsiaalid. Lihase biopsia näitab müofibrillide atroofiat. Kapillaarnekroos tekib kõigepealt piki lihaskimbu perifeeriat ja põhjustab külgnevate müofibrillide isheemiat. Kõige rohkem väljendub atroofia kimpudes, mis puutuvad kokku suurte fastsiajuhtumitega. I ja II tüüpi kiud (tooniline ja faasiline) on võrdselt mõjutatud.
Ravi.Põletikuline protsess on aktiivne 2 aastat. Kortikosteroidid vähendavad selle aktiivsust, aidates vähendada sümptomeid. Parimad tulemused saavutatakse, kui kortikosteroide manustatakse haiguse varases staadiumis, suurtes annustes ja pikka aega. Prednisoloon on valitud ravim. Selle algannus on 2 mg / kg päevas, kuid mitte suurem kui 100 mg / päevas. Kehatemperatuur normaliseerub sageli esimese 48 tunni jooksul pärast ravi algust. Mõnikord taastub CPK tase
normaalseks 2. ravinädalal paralleelselt lihaste kontraktsiooni tugevuse märgatava suurenemisega. Sellisel juhul võib prednisolooni edasist manustamist läbi viia vastavalt skeemile igal teisel päeval ja annuses, mis vähendab steroidravi kõrvaltoimete raskust. Ravi prednisolooniga on sama efektiivne, kui ravimit võetakse iga päev või graafiku alusel ülepäeviti, kuid ainult juhtudel, kui ravi ei katkestata. Kui lihasjõud suureneb, võib ülepäeviti manustatava prednisolooni algannust 5 kuu jooksul vähendada 10% kuus. Prednisolooni annuse edasine vähendamine on lubatud ainult 5% kuus. Kortikosteroidide annuse vähendamise otsustamisel on vastuvõetamatu keskenduda ainult CPK aktiivsuse vähenemisele, kuna lihasjõu märgatav kasv toimub alles 1-2 kuud pärast ensüümi taseme langust, s.o. Kortikosteroidide annuse vähendamise juhtiv kriteerium on positiivne kliiniline dünaamika. Enamikul patsientidel on prednisolooni säilitusannus, mida võetakse vastavalt skeemile ülepäeviti, mis on vajalik lihaste kontraktsioonitugevuse ja CPK kontsentratsiooni normaliseerimiseks, 25% algannusest.
Prednisooniga ravimisel kaob lööve mõnel patsiendil täielikult, kuid suurem osa jääb nahas esinevateks muutusteks. Pikaajaline steroidravi nõuab seedetrakti funktsiooni jälgimist. Mao limaskesta kaitsmiseks on ette nähtud kaaliumkloriidi ja H2-retseptorite blokaatorid. Pikaajalise ravi tõsine tüsistus on steroidse müopaatia teke, mida võib pidada põhihaiguse ägenemiseks. Kliiniliste kriteeriumide järgi on üsna raske eristada arenevat steroidset müopaatiat dermatomüosiidi ägenemisest. Steroidse müopaatia korral kannatavad reeglina proksimaalsed jäsemed, tekivad väljendunud atroofiad ja CPK aktiivsus ei suurene. Enamikul dermatomüosiidiga lastel paraneb ravi 3 kuu pärast, kuid prednisoloonravi tuleb jätkata 2 aastat. Kui ravi katkestatakse enneaegselt, on ägenemised vältimatud, tekivad lupjumine ja kontraktuurid. Narkomaaniaravile lisandub füüsiline taastusravi, vajalikud on hingamisharjutused. Massaaž aktiivses faasis on vastunäidustatud. Nõuetekohase ravi korral täheldatakse soodsat tulemust 80% -l dermatomüosiidiga lastest. Prednisooni suhtes resistentsuse või talumatuse korral
tsütostaatikumide suukaudne manustamine on näidustatud: metotreksaat annuses 10 kuni 20 mg / m 2 kehapinna kohta 2 korda nädalas või asatiopriin annuses 50-150 mg / päevas. Ravi ajal on vajalik regulaarselt jälgida maksafunktsiooni ja vererakkude koostist. Kortikosteroidide ja tsütostaatikumide kombinatsioon väldib pikaajalist ravi prednisooni suurte annustega. Juhtudel, kui kortikosteroidide kasutamist piiravad nende kõrvaltoimed, kasutatakse plasmafereesi või immunoglobuliini intravenoosset infusiooni. Mitteaktiivses staadiumis ägenemisi tavaliselt ei esine.
Polümüosiit. Enamikul juhtudel jääb etioloogia teadmata. Eeldatakse, et patogeneesis mängivad rolli rakulised ja humoraalsed mehhanismid, mida kinnitab haiguse sage areng autoimmuunprotsesside taustal (süsteemne erütematoosluupus, nodoosne periarteriit, reumatoidartriit, sklerodermia), aga ka hea toime. kortikosteroidide ja immunosupressorite kasutamisest. Patogenees on seotud rakkude poolt vahendatud tsütotoksilise reaktsiooniga, mille viivad läbi lihaskiudude pinnaantigeenide suhtes sensibiliseeritud T-lümfotsüüdid.
kliiniline pilt. Polümüosiit tekib tavaliselt täiskasvanueas (45-55 aastat), lastel ja noorukitel esineb harva ning seda ei seostata pahaloomuliste kasvajatega. Järk-järgult, järk-järgult suureneb sümmeetriline proksimaalne lihasnõrkus, palavik ja müalgia on ebatüüpilised. Sageli areneb kaela painutajate nõrkus ("rippuv pea"). Seda haigust iseloomustavad düsfaagia ja astmahood. Järk-järgult levib nõrkus distaalsetesse jäsemetesse. Pareesi raskusaste on erinev ja rasketel juhtudel areneb tetrapleegia. Mõnikord on nõrkus piiratud distaalsete lihasrühmadega, silma või näo lihastega. Patsiendil võivad esineda stabiliseerumis- ja isegi remissiooniperioodid, mis võib viia jäsemevöö müodüstroofia eksliku diagnoosini. Kroonilise haiguse käigus suureneb lihaste atroofia järk-järgult; kontraktuuride võimalik moodustumine. Kõõluste refleksid tekivad haiguse varajases staadiumis ja vähenevad lihasmassi vähenedes, kuid ei kao kunagi täielikult. See kõige olulisem diferentsiaaldiagnostiline tunnus võimaldab polüneuropaatiat välistada. Mõnikord algab haigus ägedalt üldise halb enesetunne; mõne päeva jooksul tekib terav lihasnõrkus, ilmneb valu õlavöötme lihastes. Lihaste atroofia on väga kerge
või puudub. Lihased näitavad sageli röntgenülesvõtetel lupjumisi. Täiskasvanutel on kardiopulmonaalsed tüsistused tüüpilised, mis ei ole iseloomulikud haiguse lapsepõlves esinevale vormile.
Diagnostika.CPK muutused on haruldased. EMG uuring näitab peaaegu alati nii müopaatiliste kui ka neurogeensete protsesside tüüpilisi tunnuseid. Lihase biopsia näitab mitmesuguseid patoloogilisi kõrvalekaldeid. Histoloogiliselt ei täheldata perivaskulaarset põletikulist infiltratsiooni alati, seega ei välista rakuliste infiltraatide puudumine biopsiaproovides polümüosiidi diagnoosimist.
Polümüosiidi raviks kasutatakse sama skeemi nagu dermatomüosiidi korral. Kortikosteroidide suhtes resistentsetele patsientidele määratakse tsütostaatikumid (metotreksaat). Plasmaferees ja intravenoosne immunoglobuliin on õigustatud alternatiivsed ravimeetodid, kui tavaravi ei ole piisavalt efektiivne.
Äge nakkuslik müosiit tekib pärast grippi või muud hingamisteede viirusnakkust. Viirusinfektsiooni sümptomid püsivad 1 kuni 7 päeva ja seejärel ilmnevad lihastes intensiivne sümmeetriline valu ja nõrkus. Rasketel juhtudel muutub patsient immobiliseeritud 1 päeva jooksul. Üldise nõrkuse taustal on proksimaalsed lihasrühmad tugevamalt mõjutatud kui distaalsed. Lihaste valulik palpatsioon. Kõõluste refleksid säilivad. CPK tase on tavaliselt üle 10 korra suurem kui normi ülemine piir. Peaaegu kohe pärast müosiidi tekkimist täheldatakse selle spontaanset vastupidist arengut. Halvimal juhul kulub valusündroomi kadumiseks 2–7 päeva voodipuhkust, mille järel patsient taastub täielikult.
Müotoonia.Müotoonia nähtus on lihase hilinenud lõõgastusreaktsioon pärast selle kokkutõmbumist. Määrake aktsioonimüotoonia, löökpillid või mehaaniline müotoonia ja elektromüograafiline müotoonia.
Müotoonia patogeneesis mängib rolli lihaskiudude membraani ebastabiilsus, mis põhjustab lihase korduvaid kokkutõmbeid vastuseks ühele stiimulile. Korduvad müotoonilised impulsid ei teki spontaanselt, vaid alati välismõjul või tahtliku kokkutõmbumise tulemusena. Patsiendil võib pärast lihaste intensiivset kokkutõmbumist täheldada toimemüotooniat. Patsiendil palutakse näiteks pintsel tugevalt sisse pigistada

Riis. 6.14.Müotoonilised nähtused Thomseni müotooniaga lapsel:
a- lihaste pseudohüpertroofia; b- lihaste rull müatooniga
reaktsioonid; sisse- võimetus käsi lõdvestada korduvate liigutuste ajal

Riis. 6.15.

Riis. 6.16.Thomseni müotooniaga lapse müotoonilised nähtused
rusikas ja seejärel kiiresti lahti (joon. 6.14-6.16). Sel juhul on harja täielikuks avamiseks teatud ajaline viivitus. Sama ülesande uuesti sooritamisel müotooniline nähtus iga kord väheneb ja lõpuks kaob. Kaasasündinud paramüotooniaga täheldatakse vastupidist nähtust - müotoonia suurenemist korduvate liigutustega (paradoksaalne müotoonia). Löökriistade müotoonia väljendub lihaste kokkutõmbumises pärast mehaanilist stimulatsiooni (kiire ja jõuline haamri löök lihasele). Seda nähtust võib täheldada igas lihases, kuid kõige muljetavaldavam näeb see välja siis, kui lööb lihaseid: toimub pöidla kiire painutamine ja liitmine peopesale, mis kestab mitu sekundit. Suurte lihaste löökide korral ilmnevad "rulli" ja "kraavi" sümptomid; keele põiki löökpillidega moodustub keele "konstriktsioon" või "soogu". Elektromüograafiline müotoonia registreeritakse, kui nõel süstitakse lihasesse

Riis. 6.17.EMG müotoonias, "sukeldumispommitaja droon"

Riis. 6.18.Thomseni müotoonia lapsel. "Heraklese lihased"
elektrood. Aktiivne lihaspinge või selle löök põhjustab kõrgsageduslike korduvate tühjenemiste ilmnemist, mille sagedus (100-150 Hz) ja amplituud algul suurenevad ning seejärel vähenevad. Selliste heidete kogukestus on umbes 500 ms ja heliekvivalent meenutab tuukripommitaja mürinat (joon. 6.17).
Müotoonia nähtus on mitme heterogeense päriliku haiguse kõige olulisem sümptom (joonis 6.18, 6.19).
Müotooniline düstroofia või Rossolimo-Kurshmani-Steinert-Batteni tõbi, on multisüsteemne haigus, mis pärineb autosomaalselt domineerival viisil patoloogilise geeni muutuva läbitungimisega. Haiguse etioloogia on seotud 19. kromosoomi DNA piirkonna ebastabiilsusega, mis väljendub selle patoloogilises amplifikatsioonis (korduvuses). Selle tulemusena suureneb selle geeni koopiate arv 50-lt mitme tuhandeni. Müotoonilist düstroofiat võib õigustatult omistada niinimetatud nukleotiidikolmikute laienemise haiguste klassile. Korduste arv suureneb järgnevatel põlvkondadel ja korreleerub haiguse raskema käiguga (ootusnähtus). Korduste arv lapsel

Riis. 6.19.Thomseni müotoonia täiskasvanud patsiendil

Riis. 6.20.Myotonia Rossolimo-Kurshman-Steinert-Batten. Tüüpiline patsiendi välimus
emalt haigust pärandes suureneb see palju suuremal määral kui isalt pärimisel. 100 trinukleotiidi kordusega emal on suurem kui 90% risk saada 400 kordusega laps.
See haigus on kõige levinum lihasdüstroofia tüüp, mis esineb täiskasvanutel. Haiguse esinemissagedus on 3-5 juhtu 100 000 elaniku kohta. Mõlemad sugupooled on mõjutatud võrdse sagedusega. Esimesed sümptomid ilmnevad tavaliselt teismelistel. Kaugelearenenud staadiumides täheldatakse müotooniat, näolihaste ja distaalsete jäsemete nõrkust, katarakti, frontaalset alopeetsiat, hulgi endokrinopaatiat. Näolihaste atroofia on välimuselt nii stereotüüpne, et kõik patsiendid näevad välja sarnased: nägu on pikenenud ja õhuke oimus- ja mälumislihaste nõrkuse tõttu; kael on õhuke ("luik") sternocleidomastoid lihaste atroofia tõttu; silmalaud ja suunurgad on langenud, näo alumine pool langeb, mis muudab ilme kurvaks. Jäsemete atroofia on kõige enam väljendunud distaalsetes osades: küünarvarred ja peroneaallihased (joon. 6.20, 6.21). Tekib düsfaagia, mis on tingitud neelulihaste ja söögitoru silelihaste kahjustusest. Kõõluste refleksid vähenevad ja kaovad.

Haiguse hilisemates staadiumides areneb käte väikeste lihaste atroofia. Patsiendid kaebavad lihaspinge, jäikusest tingitud liikumisraskuste üle. Müotoonia suureneb külmaga. Üldiselt ei ole müotoonilised nähtused nii väljendunud kui kaasasündinud müotoonia korral. Arst saab müotoonilise sündroomi tuvastada küsitlemisel ja kinnitada uuringul. Näiteks ei suuda müotoonilise düstroofiaga patsient kätt surudes koheselt lahti võtta. Müotoonilise düstroofia ekstraneuraalsed sümptomid - katarakt, frontaalne alopeetsia või endokriinsed häired - ilmnevad juba enne kliiniliselt olulisi müotoonia sümptomeid. Sageli registreeritakse EKG muutused. Hilisemates staadiumides võib tekkida raske kardiomüopaatia põikiblokaadiga, Adams-Stokes-Morgagni rünnakud ja südamepuudulikkus. Soole peristaltika on häiritud, areneb megakoolon. Diafragma ja interkostaalsete lihaste parees põhjustab hüpoventilatsiooni ja korduvaid bronhopulmonaarseid infektsioone. Endokriinsüsteemi häirete hulka kuuluvad munandite atroofia, naiste viljatus, hüperinsulinism, suhkurtõbi, neerupealiste atroofia ja kasvuhormooni sekretsiooni häired. Sageli tekib hüpersomnia ja obstruktiivne uneapnoe, psüühikahäired kuni raske dementsuseni.
Diagnoos põhineb iseloomulikel kliinilistel ilmingutel ja perekonna ajalool. EMG näitab müotoonilisi nähtusi, müopaatilisi potentsiaale ja kergeid denervatsiooni märke. CPK aktiivsus vastab enamasti normile. Diagnoosi kinnitamiseks ei ole vaja lihaste biopsiat. DNA analüüs tuvastab trinukleotiidi korduste arvu suurenemise; seda saab kasutada asümptomaatiliste patsientide tuvastamiseks ja sünnieelse diagnoosi tegemiseks.
Ravi.Müotoonia sümptomid nõrgenevad ravimite väljakirjutamisel - membraani stabilisaatorid: kinidiin, novokaiinamiid, fenütoiin
Riis. 6.21.Myotonia Rossolimo-Kurshman-Steinert-Batten. "Luige" kael sternocleidomastoid lihase atroofia tõttu. Küünarvarte sirutajalihaste, peroneaalsete lihasrühmade atroofia, mis põhjustab kuke kõnnaku ilmnemist
(difeniin) ja karbamasepiin (finlepsiin). Tuleb meeles pidada, et müotoonia iseenesest ei kahjusta patsienti ega vaja pidevat ravimteraapia. Paraku ei ole süveneva lihasnõrkuse ravi veel efektiivne. Patsiendid reageerivad sageli ravile negatiivselt; ei talu anesteesiat, mida võib komplitseerida pahaloomulise hüpertermia tekkega.
Kaasasündinud müotooniline düstroofia. Müotoonilise düstroofiaga emal on haiguse kaasasündinud vormiga lapse saamise tõenäosus 1:4 ja kui isa on haige - 1:12. Sünnieelse perioodi patoloogia peamised tunnused kaasasündinud vormis on loote ja polühüdramnionide motoorse aktiivsuse vähenemine. 50% lastest sünnib enneaegselt. Sünnitus võib venida emaka ebapiisava kokkutõmbumise tõttu ja sageli on vaja tange. Mõnel vastsündinul on diafragma ja roietevaheliste lihaste funktsioon nii tugevalt kahjustatud, et nad ei suuda üldse iseseisvalt hingata. Vahetu intubatsiooni ja mehaanilise ventilatsiooni puudumisel surevad paljud neist kohe. Kõige märgatavamad kliinilised sümptomid vastsündinutel on: näo dipleegia, mille puhul suu on ebatavaliselt terav ja ülahuule kuju meenutab ümberpööratud ladina tähte "V"; generaliseerunud lihaste hüpotensioon; liigese deformatsioon, mis ulatub kahepoolsest lampjalgsusest kuni laialt levinud artrogrüpoosini; seedetrakti düsfunktsioon mao lihaste pareesina, neelamis- ja aspiratsioonihäired. Nõrkus on kõige enam väljendunud proksimaalsetes jäsemetes. Kõõluste refleksid puuduvad. Müotoonilisi nähtusi ei põhjusta lihaste löökpillid ja neid ei pruugi EMG tuvastada. Vastsündinute suremus ulatub 16% -ni ja on sageli tingitud kardiomüopaatiast. Ellujäänud lastel lihasjõud reeglina suureneb ning toitumis- ja hingamisprotsessid normaliseeruvad 1 elukuu jooksul.
Pikaajaline prognoos on ebasoodne: vaimne alaareng ja müotoonilise düstroofia väljendunud kliinilised sümptomid on leitud kõigil lastel. Diagnoosimiseks on vaja diagnoosida müotooniline düstroofia emal, kellel on tavaliselt mitmeid haiguse kliinilisi tunnuseid ja müotoonilisi EMG nähtusi.
Ema ja lapse diagnoosi saab täpsustada pärast 19. kromosoomi DNA segmendi amplifitseerimist. Pereliikmed on ohus ja seejärel läbivad kandmise kindlakstegemiseks geneetilise testimise.
Vastsündinu kiirabi koosneb kohesest intubatsioonist ja mehaanilisest ventilatsioonist. Seedetrakti funktsioon normaliseerub cerukaali (metoklopramiidi) määramisega. Kasutamisel väheneb liigeste jäikus füüsilised meetodid teraapia ja immobiliseerimine.
kaasasündinud müotoonia - pärilik haigus, mida iseloomustab jäikus ja tõeline lihaste hüpertroofia. 19% peredest tuvastatakse autosoomne dominantne pärand (Thomseni tõbi), harvem - autosoomne retsessiivne pärand (Beckeri tõbi). Enamik juhtumeid on juhuslikud. Üldiselt algab autosomaalse retsessiivse vormiga patsientidel haigus hiljem ja kulgeb raskemate müotooniliste häiretega kui autosomaalse domineeriva vormiga. Kuid mõlema vormi sümptomid on samad, mistõttu on võimatu teha järeldusi pärilikkuse tüübi kohta ainult kliiniliste kriteeriumide põhjal (vt joonis 6.18, 6.19).
Kaasasündinud müotoonia domineeriva ja retsessiivse vormi patoloogiline geen on kaardistatud 7. kromosoomi pikal käel, kus asub kloriidioonikanali geen.
Autosoomne dominantne vorm debüteerib tavaliselt imikueas häälemuutusega koos nutmisega; laps hakkab lämbuma ja pärast nutmist nägu lõdvestub väga aeglaselt. Haigus on kerge. Täiskasvanueas võib müotoonia viia generaliseerumiseni lihaste hüpertroofia(sportlikkus), kuid isegi lapsepõlves näevad lihased välja nagu "Heraklese lihased". Mõnikord on haaratud keele-, näo- ja mälumislihased. Lihaste jäikusega ei kaasne valu; see suureneb, kui patsient jääb külma. Selguvad löökpillide müotoonilised sümptomid. Lihasmass, kontraktsioonide tugevus ja kõõluste refleksid on normaalsed. Vahetult pärast puhkust jäävad lihased kinni ja liigutused on rasked. Pärast aktiveerimist aga jäikus kaob, taastub normaalne liikumisulatus.
Diagnostika.Diagnoosi kinnitab EMG uuring. Lihase korduvate võngete sagedus varieerub 20 kuni 80 tsüklit sekundis nõela esmasest lihasesse sisestamise hetkest kuni tahtliku kontraktsiooni alguseni. Potentsiaalide amplituud ja sagedus tõusevad ja langevad, millega kaasneb iseloomulik heli – "sukelduva pommitaja mürin". Lihasdüstroofia nähud puuduvad. CPK tase on normaalne. Lihasbiopsia proovid näitavad lihaskiudude hüpertroofiat.
Ravi.Müotoonia ei vaja alati ravi ja ravimid ei ole piisavalt tõhusad. Jäikust saab mõnikord leevendada fenütoiini (difeniini) või karbamasepiini (Finlepsin) preparaatidega, mida manustatakse mõõdukates krambivastastes annustes. Novokaiinamiid määratakse algannuses 200 mg 2 korda päevas ja seejärel suurendatakse seda järk-järgult 400 mg-ni 3 korda päevas. Ravim vähendab lihaste jäikust haiguse retsessiivse vormiga lastel. Diakarb (atsetasoolamiid) on mõne patsiendi jaoks efektiivne. Rasketel juhtudel on näidustatud lühike kortikosteroidide kuur. Kasulikud kaltsiumi antagonistid (nifedipiin 10-20 mg 3 korda päevas), samuti disopüramiid 100-200 mg 3 korda päevas. Tuleb arvestada, et suktsinüülkoliin, veroshpiroon, kaalium, antihüperlipideemilised ained ja β-blokaatorid võivad müotoonilist sündroomi süvendada.
Korduv müotoonia (müotoonia, mida süvendab liigne kaaliumisisaldus) on autosoomne dominantne sündroom, mis on seotud naatriumikanali geeni mutatsiooniga. Geen on kaardistatud kromosoomil 17. Kliinilised ilmingud on sarnased kaasasündinud müotooniaga. Lihasjäikus tekib tavaliselt pärast 10. eluaastat ja selle võib vallandada üldanesteesia. Müotoonilised nähtused on üldistatud, hõlmates kehatüve, jäsemeid ja okulomotoorseid lihaseid. Müotoonia raskusaste on päevade lõikes erinev ja väheneb soojenemisel. Seisund võib halveneda pärast intensiivset treeningut või suures koguses kaaliumisisaldust toiduga.
Diagnostika.EMG-uuring paljastab müotoonilisi nähtusi. Lihase biopsia proovides pole patoloogiat. Naatriumikanali α-subühikut kodeeriva mutantse geeni võimalik DNA analüüs.
Ravi.Müotoonia ägenemise jäikust saab ära hoida meksiletiiniga, mis on lidokaiiniga sarnase struktuuriga ravim; nagu ka teiste kanalopaatiate puhul, võib diakarb (atsetasoolamiid) olla efektiivne.
6.6. Perioodiline halvatus
Perioodiline halvatus ehk paroksüsmaalne müopleegia on katustermin kanalopaatiate rühma jaoks, harvaesinevad pärilikud haigused, mida iseloomustavad ioonkanali patoloogiast tingitud skeletilihaste lõtv halvatus. Halvatus jaguneb sõltuvalt kaaliumi tasemest veres: hüperkaleemiline (Gamstorpi tõbi), hüpokaleemiline ja normokaleemiline. Lisaks võib perioodiline halvatus
olla esmane (geneetiliselt määratud) või sekundaarne. Sekundaarne hüpokaleemiline perioodiline halvatus on põhjustatud kaaliumi kadumisest uriinist või selle liigsest eritumisest seedetraktist. Kaaliumi kadu uriiniga on seotud primaarse hüperaldosteronismi, lagritsa (lagritsa) mürgistuse, amfoteritsiin B-ravi ja mõnede neerutorude defektidega. "Seedetrakti" kaaliumikadusid täheldatakse kõige sagedamini raske kroonilise kõhulahtisuse, pikaajalise sondiga toitmise ja gastrofistuli korral. Kaalium kaob anorexia nervosaga noorukitel, kes kuritarvitavad diureetikume või oksendavad, et kaalust alla võtta. Hüpokaleemiline perioodiline halvatus raskendab türeotoksikoosi. Sekundaarne hüperkaleemiline perioodiline halvatus võib olla tingitud neeru- või neerupealiste puudulikkusest.
Perekondlik hüperkaleemiline halvatus päritud autosoomselt domineerival viisil ja kõrge penetrantsusega. Mutatsioon asub naatriumikanali geenis.
kliiniline pilt. Lihasnõrkuse rünnakute algus viitab varasele lapsepõlvele ja isegi lapseeale. Nõrkusehood tekivad pärast intensiivset füüsilist pingutust. Enne rünnakut on tundlikud häired - paresteesia näol, üla- ja alajäsemetel, raskustunne seljas. Mõnikord võib patsient kõndides või ühest kohast teise liikudes aeglustada halvatuse teket. Imikutel ja väikelastel väljenduvad rünnakud lihastoonuse järsu langusega: nad kukuvad ega saa liikuda. Vanematel lastel ja täiskasvanutel võib seda täheldada krambihoogudena mõõdukas(kestab vähem kui tund ja ei põhjusta sügavat halvatust) ja rasked rünnakud (kuni mitu tundi). Pärast mitut rasket rünnakut võib mõni järelejäänud lihasnõrkus püsida. Hüperkaleemilise halvatusega patsientidel on müotoonia sümptomid mõõdukad ja võivad jahtudes süveneda. Iseloomustab silmalaugude, keele, küünarvarre ja pöidla lihaste müotoonia.
Diagnostika.Rünnaku ajal ületab kaaliumisisaldus veres tavaliselt 5 mmol / l. Kaaliumkloriidi suukaudne tarbimine kohe pärast treeningut kutsub esile nõrkusehoo, mille käigus lihased ei reageeri elektrilistele stiimulitele.
Ravi.Ägedad rünnakud vajavad harva ravi, kuna need on lühiajalised. Mis kasutusele võetud rünnak võib aidata intravenoosselt
40% glükoosilahuse (kuni 40 ml) või 10% kaltsiumglükonaadi lahuse (kuni 20 ml) infusioon. Diakarbi (atsetasoolamiidi) igapäevane tarbimine hoiab ära korduvad rünnakud, selle ravimi ennetava toime mehhanism hüperkaleemilise ja hüpokaleemilise halvatuse korral ei ole teada. Vältida tuleks kaaliumirikaste toitude söömist, suurendada süsivesikute ja soola hulka igapäevases toidus.
Perekondlik hüpokaleemiline halvatus päritud autosomaalselt domineerival viisil. Geeni tungimine naistel on vähenenud. Mutatsioon asub 7. kromosoomi pikal käel kaltsiumikanali geenis. 60% patsientidest ilmnevad sümptomid enne 16. eluaastat, ülejäänud - kuni 20 eluaastat. Alguses on nõrkushood harvad, kuid siis on neid kuni mitu korda nädalas. Krambid provotseerivad: puhka pärast füüsilist aktiivsust (sageli täheldatakse krampe Varahommik), süsivesikuterikaste toitude rikkalik tarbimine, liigne soolasisaldus toidus, emotsionaalne stress, alkoholi tarbimine, hüpotermia; naistel - menstruatsioon. Enne rünnakut ja selle ajal võib patsiendil tekkida janu ja oliguuria, valu proksimaalsetes lihasrühmades, seejärel tekib üldine nõrkus. Mõnikord esineb täielik halvatus, mille korral patsient ei suuda isegi pead tõsta. Näolihaste nõrkust esineb harva, silmade liigutused on alati säilinud. Hingamispuudulikkus ei arene. Enamik rünnakuid kestab 6 kuni 12 tundi ja mõned - kogu päeva (nn müopleegiline seisund). Lihasjõud taastub kiiresti, kuid pärast mitut rasket rünnakut võib täheldada väsimust, kehakaalu langust, eriti proksimaalsete jäsemete puhul, ja kõõluste reflekside allasurumist. Tüüpilised on autonoomsed häired: naha punetus, liighigistamine, pulsi labiilsus ja vererõhk. Väljaspool lihasnõrkuse rünnakuid ei esine patsientidel neuromuskulaarse patoloogia sümptomeid.
Diagnostika.Rünnaku ajal võib kaaliumi tase veres langeda 1,5 mmol / l-ni, mis vastab EKG muutustele: bradükardia, laine lamenemine. T, intervallide suurenemine P-Q ja Q-T. Lihased ei tõmbu kokku vastuseks elektrilistele stiimulitele. Diagnostilistel eesmärkidel võib rünnaku esile kutsuda glükoosi võtmisega annuses 2 g / kg ja samaaegse 10-20 ühiku insuliini subkutaanse manustamisega: paralüüsihoog areneb 2-3 tunni pärast.
Ravi.Piisava neerufunktsiooniga patsientide ägedaid haigushooge ravitakse kaaliumi korduvate annustega annuses 5–10 g.
Nende esinemise vältimiseks on soovitatav võtta iga päev sama annus. Noorematel lastel on annus väiksem. Diakarbi (atsetasoolamiidi) igapäevane tarbimine on osutunud paljudel juhtudel kasulikuks krambihoogude ennetamisel. Sellel on madal toksilisus ja see on üldiselt hästi talutav isegi pikaajalisel kasutamisel. Peaks vähendama kaloreid päevane ratsioon süsivesikute arvelt ja vähendada lauasoola kogust. Samal ajal näidatakse kaaliumirikkaid toite: kuivatatud puuviljad, kuivatatud aprikoosid, ploomid, piimatooted, kartul.
Perekondlik normalemichesky halvatus. Mõnes perekonnas esineb autosomaalse domineeriva perioodilise halvatuse juhtumeid normaalse kaaliumisisaldusega veres. See on hüperkaleemilise perioodilise halvatuse variant, millega kaasneb kaaliumi verre sissevoolu rikkumine, kui selle tegelikku sisaldust kudedes ei ole võimalik hinnata. Müopleegia kestab mitu päeva kuni 2-3 nädalat. Lihasnõrkuse suurenemise ja vähenemise kiirus on tavaliselt aeglane. Kõõluste refleksid rünnakute ajal kaovad. Mõnel patsiendil täheldatakse üksikute lihasrühmade hüpertroofiat. Rünnakuid kutsub esile puhkamine pärast intensiivset füüsilist aktiivsust, alkoholi tarbimine, jahutamine. Kaaliumkloriidi võtmine võib esile kutsuda halvatuse, samas kui igapäevane 8-10 g lauasoola kasutamine väldib neid.
6.7. myasthenia gravis
myasthenia gravis(myasthenia gravis)- autoimmuunne neuromuskulaarne haigus, mida iseloomustab kliiniliselt patoloogiline nõrkus ja väsimus vabatahtlikud lihased ja on seotud vöötlihaste postsünaptilise membraani atsetüülkoliini retseptorite (ACh-R) kahjustusega spetsiifiliste komplementi fikseerivate antikehade (AT) poolt.
Myasthenia gravis'e levimus on kõigis populatsioonides 0,5-5 juhtu 100 000 elaniku kohta. Alla 17-aastased lapsed ja noorukid moodustavad 9-15% myasthenia gravis'ega patsientide arvust. Keskmine haigestumise vanus oli 7,2 aastat. Myasthenia gravise debüüt on võimalik igas vanuses. Kirjeldatakse kaasasündinud vorme. Naised haigestuvad 3 korda sagedamini kui mehed.
Etioloogia.Multifaktoriaalne haigus, mille puhul pärilik eelsoodumus on tingitud immunoloogilisest defektist ja on seotud anti-
HLA süsteemi B8 histo-sobivuse geenid. Müasteenia põhjuseks võib olla harknääre viiruslik kahjustus, mille tagajärjel hakkab see tootma muutunud membraanistruktuuriga T-lümfotsüüte; harknääre kasvaja; harvadel juhtudel erineva etioloogiaga esmane ajukahjustus.
Myasthenia gravise patogeneesi aluseks on autoimmuunne reaktsioon skeletilihaste atsetüülkoliinesteraasi retseptoritele (ACh-R). ACh-R antikehade tase patsientide veres on korrelatsioonis haiguse tõsidusega. ACh-R vastased antikehad blokeerivad neuromuskulaarset juhtivust, kuna hävitavad ACh, vähendavad selle taastumise kiirust, muutes pöördumatult postsünaptilise membraani retseptoreid.
Patoloogiline anatoomia. aksoni otstes tekivad düstroofsed muutused, sünaptilised lõhed ja postsünaptilised struktuurid, ladestavad nad immunoglobuliine ja komplementeerivad. Lihastes täheldatakse mõõdukat degeneratiivset atroofiat, harvem kiu nekroosi koos kerge lümfoidse infiltratsiooni ja plasmorraagiaga. 70-90% patsientidest avastatakse harknääre patoloogia (idude folliikulite hüperplaasia, lümfoepiteliaalsed tümoomid). Harvadel juhtudel täheldatakse müokardiiti, türeoidiiti, lümfotsüütide fokaalset kogunemist erinevates organites.
Myasthenia gravise kliiniline klassifikatsioon (vastavalt B.M. Gekhtile).
1. Liikumishäirete üldistusaste:
1) üldistatud;
2) kohalik:
a) silm
b) pirn,
c) luustik.
2. Liikumishäirete raskusaste:
1) valgus;
2) keskmine;
3) raske.
3. Müasteenilise protsessi kulg:
1) ägenemine (müasteenilised episoodid);
2) mitteprogresseeruv (müasteeniline seisund);
3) progressiivne;
4) pahaloomuline.
4. Liikumishäirete kompenseerimise määr antikoliinesteraasi ravimite mõjul:
1) täis (kuni töövõime taastamiseni);
2) mittetäielik (taastub iseteeninduse võime);
3) halb (patsiendid vajavad välisabi). kliiniline pilt. Myasthenia gravis't iseloomustab patoloogiline
vöötlihaste väsimus ja nõrkus. Patsientidel on raske trepist ronida, kõndida, pikka aega ühes asendis püsida, raskusi kanda.
Kõige sagedamini haigestuvad silma-, näo-, närimislihased, aga ka neelu-, kõri- ja keelelihased. Silma välislihaste kahjustused avastatakse esimese läbivaatuse ajal 40-50% patsientidest ja haiguse arenedes - 90-95%. Ptoos võib olla ühepoolne ja esineb ühel või teisel küljel. Hommikul ja pärast puhkust on ptoos vähem, suureneb üldise või visuaalse stressiga, õhtu poole. Uurimisel on võimalik provotseerida ptoosi suurenemist, paludes patsiendil mitu korda silmad sulgeda või istuda. Silma motoorsed häired on asümmeetrilised, koormuse all muutlikud ega vasta silmamotoorsete närvide innervatsioonitsoonidele. Lihasnõrkuse tõttu tekib nüstagm äärmuslikes juhtmetes. Diploopia suureneb visuaalse ja kehalise aktiivsuse, ereda valguse korral pärastlõunal (eriti telerit vaadates), kaugusesse vaadates väljendub rohkem, väheneb pärast puhkamist. silmad kinni ja hommikul (joon. 6.22).
Närimis- ja oimuslihaste nõrkus toob kaasa närimisel väsimuse, mõnikord alalõua lõtvumise, patsiendid toetavad söömise ajal lõualuu ja aitavad ennast kätega närides. Oluline sümptom on näolihaste nõrkus. See on rohkem väljendunud näo ülemises pooles (silmade ringlihastes), suureneb korduva kissitamise ja üldise kehalise aktiivsusega. Patsiendil on raske põski välja paisutada, nõrkuse tõttu tekib "rist" naeratus ringikujuline lihas suu. Täheldatakse ka mälumis- ja oimulihaste nõrkust.

Riis. 6.22.Silma lihaste nõrkus myasthenia gravis'e korral
Sibulalihaste (pehmesuulae, neelu ja ülemise söögitoru lihaste) kahjustused, mis põhjustavad düsfaagia ja düsartria, arenevad 40% patsientidest. See suureneb kõne, üldise kehalise aktiivsusega, söögi ajal ja väheneb pärast puhkust. Neelamine on häiritud (patsient lämbub söömisel, vedel toit satub ninakäikudesse). Kõne muutub nasaalseks, võib täheldada hääle kähedust või kogelemisele sarnaseid modulatsioonihäireid. Raske düsartria korral ei saa patsient neelata ega rääkida.
Kaela ja kehatüve lihaste nõrkus on tüüpilisem eakatele patsientidele. Seljalihaste nõrkus väljendub kehahoiaku rikkumises. Kaela tagumise lihasrühma nõrkuse tõttu muutub pea tõstmine lamavas asendis või kaela sirutamisel raskeks. vertikaalne asend. Kui myasthenia gravis debüteerib koos kehatüve lihaste nõrkusega, tekivad tulevikus bulbar- ja hingamishäired.
Kaebused õhupuuduse kohta sissehingamisel on tingitud diafragma või roietevaheliste lihaste nõrkusest. Köhašoki nõrgenemine toob kaasa paksu röga, viskoosse sülje kogunemise, mida ei saa välja sülitada ega alla neelata.
Jäsemete, eriti proksimaalse, kaela ja kehatüve lihased on nõrgenenud. Uurimisel lihasatroofia, lihastoonuse langus, kõõluste labiilsus ja periosteaalsed refleksid. Jäsemete lihaste nõrkust võib isoleerida (ilma muude müasteenia sümptomiteta) või kombineerida teiste lihasrühmade nõrkusega. Tüüpiline on proksimaalsete sirutajalihaste nõrkus. Kõige sagedamini on kahjustatud deltalihas, õla triitseps ja niudelihas.
Lisaks motoorsete häiretele kaasnevad müasteeniaga mitmesugused autonoomsed ja endokriinsed häired(hüpo- ja hüpertüreoidism, hüpokortisism jne). Myasthenia gravis't iseloomustab lihasnõrkuse dünaamilisus päeva jooksul, selle tugevnemine pärast treeningut, pöörduvus või nõrkuse vähenemine pärast puhkust. Halvenemist provotseerivad füüsiline aktiivsus, negatiivsed emotsioonid, menstruatsioon, infektsioonid, ümbritseva õhu temperatuuri tõus ja paraneb - ööuni, lõõgastus. Väsimuse vähenemine pärast antikoliinesteraasi ravimite (ACP) manustamist on patognoomiline.
Haiguse kulg on enamasti progresseeruv, remissioonidega või progresseeruv ilma remissioonideta. Pahaloomulise kulgemise korral tekivad haiguse esimestel nädalatel bulbar- ja hingamishäired. Müasteenia debüteerib sageli pärast SARSi või
stress, üks sümptom (mööduv ptoos, bulbar parees jne). Müasteeniaga patsientide seisundit võivad komplitseerida müasteenilised kriisid või kolinergilised kriisid.
müasteeniline kriis areneb myasthenia gravise dekompensatsiooni või ACP ebapiisava annuse tõttu; võib olla põhjustatud bronhopulmonaarsest infektsioonist. Sel juhul on seisund järsult halvenenud koos elutähtsate funktsioonide rikkumisega. Müasteenilist kriisi saab eristada teistest rasketest seisunditest, millega kaasnevad hingamishäired, asümmeetrilise välise oftalmopareesi, ptoosi, bulbarsündroomi, hüpomia, jäsemete ja kaela lihaste nõrkuse, mis väheneb vastusena AChE ravimite manustamisele (tabel). 10).
Kolinergiline kriis areneb AChE ravimite ülemäärase annusega.
Tabel 10Müasteeniliste ja kolinergiliste kriiside diferentsiaaldiagnostika

Segatud (müasteenilised + kolinergilised) kriisid esineb myasthenia gravis'ega patsientidel, kellel on ebaõige tarbimine ja/või algselt kitsas vahemik terapeutilised annused AHEP, samuti seisundite taustal, mis põhjustavad üldist või lihaste nõrkus erineva päritoluga (kaasuvad infektsioonid, somaatilised, hormonaalsed häired, ravimite võtmine, mis mõjutavad vabatahtlike lihaste kontraktiilset funktsiooni jne).
Prognoos sõltub kliinilisest vormist ja ravist. Võimalik on praktiline paranemine (umbes 1/3 patsientidest), märkimisväärne paranemine, puue, surmad, eriti tümoomiga. Peamised sümptomid, mis ohustavad patsiendi elu, on kõri- ja hingamislihaste nõrkus. Müasteenia surma põhjused: hingamispuudulikkus, aspiratsioonipneumoonia, kortikosteroidide ja tsütotoksiliste ravimite kõrvaltoimed.
Diagnoos hõlmab anamneesi kogumist, kliinilist läbivaatust, testi AChE preparaatidega (prozeriin, tensilon, kalymin), elektromüograafiat, immunoloogilist uuringut, harknääre uuringut, lihasbiopsia morfoloogilist uuringut, dünaamilist vaatlust.
Kliiniline läbivaatus hõlmab üldise neuroloogilise seisundi uuringut ning näo-, kaela-, kehatüve- ja jäsemete vabatahtlike lihaste tugevuse hindamist enne ja pärast treeningut. Lihasjõudu hinnatakse 0–5 punkti, kus 0 on jõudu puudumine, 5 on normaalne tugevus, arvestades vanust ja sugu. Samuti tuvastatakse kesknärvisüsteemi kahjustuse sümptomite puudumisel lihaste patoloogilise väsimuse sündroom (sümptomite suurenemine pärast treeningut).
Diagnostilised kriteeriumid
1. Ptoos (ühepoolne, kahepoolne, asümmeetriline, sümmeetriline): ptoosi ilmnemine või intensiivistumine pärast pikka aega üles vaatamist või pärast silmade kiiret avamist või sulgemist korduvalt.
2. Närimislihaste nõrkus:
Ebapiisav vastupidavus alalõualuu sunnitud sulgemisele;
Ajutiste lihaste palpatsioon närimise ajal näitab nende nõrka kontraktsiooni;
Patsiendid ei suuda silmalaugusid tihedalt sulgeda ega takistada silmade passiivset avanemist;
Patsiendid ei saa neile vajutades põski täis puhuda.
3. Kõri- ja suulaelihaste nõrkus avastatakse, kui:
Suulae on passiivne, oksendamise refleks on vähenenud või puudub;
Raskused vedela toidu neelamisel.
4. Keelelihaste nõrkus avastatakse, kui keel surutakse läbi põse arsti sõrmele.
5. Kaelalihaste tugeva nõrkusega “pea ripub alla”.
6. Proseriini test koos lihasjõu ja väsimuse hindamisega viiakse läbi enne 0,05% proseriini lahuse subkutaanset süstimist ühekordse vanuseannusena ja 30-40 minutit pärast seda. Test loetakse positiivseks, kui lihasjõud suureneb. Eristama:
Teravalt positiivne test, kui kõik müasteenilised sümptomid kaovad;
Positiivne test - jäävad ainult üksikud sümptomid;
Nõrgalt positiivne test, mille puhul müasteeniliste sümptomite raskusaste väheneb;
Kahtlane proseriini test - myasthenia gravise ilmingute raskus muutub veidi;
Prozeriini test negatiivne - kliinilised sümptomid ei muutu pärast prozeriini kasutuselevõttu.
Myasthenia gravise diagnoosi kinnitamine on prozeriini testi ühe esimese kolme variandi olemasolu.
Neuromuskulaarsete ülekandehäirete (lihas, mis eemaldab suu põhja digastrilise lihase väikese sõrme) tunnuste tuvastamiseks tehakse kõige nõrgemate lihaste EMG. Uuring viiakse läbi AHEP kaotamise taustal päeva jooksul, vahetult pärast treeningut ja 2 minutit pärast treeningut. Väga oluline on EMG nähtuste pöörduvus ACEP taustal - M-vastuse amplituudi suurenemine. Elektromüograafia näitab teise lihase aktsioonipotentsiaali amplituudi vähenemist (tavaliselt on mõlemad potentsiaalid võrdsed) vastuseks närvistimulatsioonile paarisimpulssidega intervalliga 0,1-0,7 s. Müasteenia korral asendub potentsiaalide amplituudi vähenemine närvi pideva stimulatsiooniga platoofaasi või amplituudi suurenemisega ning teiste haiguste korral toimub reaktsiooni amplituudi pidev vähenemine. Üksikute lihaskiudude aktiivsuse registreerimisel ilmnevad sageli neuromuskulaarsete sünapside kahjustuse tunnused. 95% juhtudest tuvastab EMG patognoomilisi muutusi.
Harknääre kasvaja või hüperplaasia välistamiseks, mis areneb 75% -l myasthenia gravis'ega patsientidest, tehakse mediastiinumi kompuutertomograafia ja radionukliidide skaneerimine.
Immunoloogiline uuring näitab kolinergiliste retseptorite vastaste antikehade olemasolu 50% -l myasthenia gravis'e okulaarse vormiga patsientidest ja 80-90% -l üldistatud vormiga patsientidest. Tümoomiga tuvastatakse ka skeletilihaste antigeenid.
Immunoloogiline uuring (ELISA, RIA) on kvantitatiivne meetod AChR-i antikehade määramiseks myasthenia gravis'ega patsientide vereseerumis, mis võimaldab diagnoosi kinnitada kuni 80% tõenäosusega.
Diferentsiaaldiagnostika viiakse läbi haigusseisunditega, mille peamiseks sümptomiks on lihasnõrkus:
Müasteenilised sündroomid (botulism, mürgistus aminoglükosiidide rühma antibiootikumidega, Itsenko-Cushingi tõbi, Addisoni tõbi, hüpo- ja hüpertüreoidism, poliomüosiit);
Sclerosis multiplex, neuroinfektsioonid (entsefaliit, polüneuropaatia, entsefalomüelopolüradikuloneuriit): patsientidel kaasneb oftalmopareesiga hüporefleksia, ataksia, tundlikkuse häired, tserebrospinaalvedeliku muutused;
Amüotroofne lateraalskleroos: pidev nõrkus, atroofia, fastsikulatsioonid, suurenenud kõõluste refleksid, Babinsky sümptom;
Müopaatia okulaarne vorm: iseloomulikud on ptoos ja silmamuna liigutuste sümmeetriline piiramine; neelu, kaela, jäsemete ja näo lihaste kerge nõrkus;
mitokondriaalsed müopaatiad;
neuroendokriinsed sündroomid;
Teised kesknärvisüsteemi haigused (kasvajad, pea- ja seljaaju veresoonte haigused): iseloomulikud on refleksihäired, juhtivuse häired;
Astenoneurootilised reaktsioonid, kroonilise väsimuse sündroom jne.
Ravi.Üldised põhimõtted:
1. Generaliseerunud vormiga patsient hospitaliseeritakse ja kehaline aktiivsus on piiratud kuni antikoliinesteraasi ravi valimiseni.
2. Neuromuskulaarset ülekannet blokeerivad ja kesknärvisüsteemi pärssivad vahendid on vastunäidustatud;
ja eriti hingamiskeskusele (kiniin, kinidiin, propranolool, lidokaiin, aminoglükosiidid, polümüksiin, morfiin, barbituraadid, rahustid). 3. Ravi eesmärgid sõltuvad haiguse tõsidusest. Antikoliinesteraasi ravimid (ACEP)- müasteenia jaoks valitud ravimid, mis pärsivad atsetüülkoliini hävimist ja aitavad kaasa selle akumuleerumisele sünaptilises pilus, toimides kolinergilistes sünapsides, ei tungi BBB-sse (tabel 11). Kõrvaltoimed tulenevad samaaegsest toimest autonoomsele kolinergilisele sünapsile ja sõltuvad ANS-i annusest ja toonist. Neid saab vähendada, kui AChE inhibiitoreid võtta sagedamini, kuid väiksemates annustes ja koos toiduga, mis aeglustab imendumist. Mõnes olukorras (menstruatsioon, infektsioonid, remissioon) suureneb tundlikkus AHEP suhtes ja nende annust vähendatakse. Patsiente õpetatakse annust ise kohandama. AHEP kasutamise suhtelised vastunäidustused on bronhiaalastma, raskekujuline ateroskleroos, pärgarteritõbi, epilepsia.
Tabel 11Antikolinergilised ravimid
Ettevalmistused | Tegevuse aeg | Kasutusvaldkonnad |
Prozerin (neostigmiin) | Tegevuse algus 20-40 minuti pärast, kestus 2-4 h | Seda kasutatakse peamiselt uimastitestimiseks ja ägedate seisundite korral. |
Kalimin 60 N, kalimin-forte (püridostigmiinbromiid) | Algab 45 minuti pärast, kehtib 4-8 h Annuste vaheline intervall on 5-5,5 tundi. | Kõige laialdasemalt kasutatav, hästi talutav, efektiivne kõigis vormides, sealhulgas bulbar. Kalimin forte (parenteraalne) - elutähtsate funktsioonide rikkumise ja püsiva bulbaarse halvatuse korral. Patsientide üleviimisel ravimite parenteraalsele manustamisele võetakse arvesse, et 1 tablett kaliminat (60 mg) võrdub 1 ml 0,05% prozeriini lahusega. |
Täiendav teraapia: kaaliumipreparaadid (pikendavad AHEP toimet); kaaliumirikas dieet (küpsetatud kartulid, kuivatatud aprikoosid, banaanid jne); kaaliumi säästvad ravimid (veroshpiron); kaaliumkloriid 3,0 g/päevas lahuste, pulbrite, tablettidena AHEP üleannustamise vältimiseks; kaltsiumipreparaadid; toonik (eleutherococcus, Rhodiola, Leuzea, pantocrine ekstraktid); multivitamiinid, aminofülliin (fosfodiesteraasi blokaator, mis suurendab cAMP sisaldust presünaptilises membraanis), anaboolsed ained (riboksiin, retaboliil).
Patogeneetiline teraapia - tümektoomia. Efektiivsus - 70-90%, remissioonid on võimalikud. Kirurgilise ravi näidustused on järgmised:
a) myasthenia gravise pahaloomulised vormid;
b) myasthenia gravise progresseeruv vorm;
c) müasteeniline seisund, olenevalt defekti raskusastmest.
Tümektoomia vastunäidustused:
a) rasked dekompenseeritud somaatilised haigused;
b) vanadus.
Preoperatiivne ettevalmistus hõlmab taastavat ravi, plasmafereesi, vastavalt näidustustele - glükokortikoidid, kiiritusravi (vastunäidustatud lastel ja noorukitel).
Glükokortikoidid (prednisoloon, deksametasoon) näidatakse, kui muud meetodid on ebaefektiivsed. Neid määratakse iga päev või ülepäeviti, 60-150 mg / päevas (1-1,5 mg / kg / päevas) hommikul, kohe pärast hommikusööki, ülepäeviti; väljendunud ägenemisega, iga päev (kuni elutähtsate häirete kompenseerimiseni), 5-7 päeva pärast (kuni ravitoime ilmnemiseni) lülituvad nad skeemile üle ülepäeviti. Säilitusannus - ülepäeviti 20-30 mg päevas, võetakse mitu kuud. Ligikaudu 75% patsientidest toob kortikosteroidravi kaasa märkimisväärse paranemise. Pärast stabiilset paranemist vähendatakse kortikosteroidide annust aeglaselt (mitme kuu jooksul) säilitusravini (5-15 mg päevas või 10-30 mg ülepäeviti). Mõnikord on võimalik kortikosteroide täielikult tühistada. Esialgse halvenemise vältimiseks võib ravi alustada väikeste annustega (25 mg prednisolooni ülepäeviti), suurendades järk-järgult annust 12,5 mg võrra iga kolmanda annuse järel, kuni päevane annus jõuab 100 mg-ni või saavutatakse hea toime. Paranemist täheldatakse pärast 6-7-nädalast ravi. Nendel juhtudel hakatakse annust vähendama mitte varem kui 3 kuud pärast esimest annust.
Plasmafereesette nähtud ägenemiste, müasteeniliste kriiside, operatsioonieelse ettevalmistuse, kortikosteroidravi ebaefektiivsuse korral. Ülepäeviti tehakse 3-5 seanssi, seejärel 2-3 korda nädalas. Plasmaferees viiakse läbi plasmavahetuse või valguasendajate kasutamisega. Hemosorptsioon ja enterosorptsioon viiakse läbi myasthenia gravis'e generaliseerunud vormiga patsientidel antikehade eemaldamiseks ning segakriiside ja massiivse ravimteraapia ebaefektiivsuse korral - detoksifitseerimiseks.
Tsütostaatikumid (asatiopriin, tsüklofosfamiid ja tsüklosporiin) määratud vereanalüüside kontrolli all. Immunoglobuliin G preparaadid (in / in / in / in 0,4 g / kg / päevas 5 päeva jooksul või 3-5 g ühe kuuri kohta) on efektiivsed kaasnevate infektsioonide korral, müasteenilise või segakriisi ajal.
KriisiraviSee on suunatud elutähtsate häirete kompenseerimisele, ägenemise leevendamisele ja ainevahetushäirete kõrvaldamisele. Müasteenilise kriisi ravis manustatakse AHEP-d parenteraalselt (kalimin-forte 1-1,5 ml IV või IM iga 4-5 tunni järel või prozeriin 1,5-2 ml iga 3 tunni järel). ALV koos AHEP täieliku kaotamisega määratakse antibakteriaalsete ravimite taustal immunosupressiivne ravi, et vältida vahelduvaid infektsioone. Seadmest lahtiühendamine toimub alles pärast 30-minutilist spontaanset hingamist koos hingamishäirete kompenseerimisega ja kalimina-forte taustal 5-6 tundi.Suured glükokortikoidide annused määratakse vahelduva skeemi järgi (impulssteraapia - 1000). -2000 mg IV tilguti igal teisel päeval), millele järgneb üleviimine suukaudsele u. Nad stabiliseerivad ka kardiopulmonaalset aktiivsust. Tehakse plasmaferees, normaalse inimese immunoglobuliini intravenoossed infusioonid. Kolinergilise kriisi peatavad atropiin, koliinesteraasi reaktivaatorid (dipiroksiim); kasutada võõrutusravi.